Themen • Kurzbeiträge • Streiflichter
Ursprung der Bakterien – aktuelle Versuche „evolutionäre Bäume“ zu retten
von Boris Schmidtgall
Studium Integrale Journal
28. Jahrgang / Heft 2 - September 2021
Seite 116 - 120
Zusammenfassung: Das Aufstellen von Stammbäumen der Lebewesen ist ein wichtiges Element der evolutionstheoretisch motivierten Forschung. Kürzlich haben zwei wissenschaftliche Gruppen von Fortschritten auf der Suche nach dem letzten gemeinsamen Vorfahren aller Bakterien berichtet. Beide Gruppen führten hierzu phylogenetische Analysen an einer Vielzahl bakterieller Genome durch. Es wird deutlich, dass die Ergebnisse viel mehr von den gewählten Prämissen abhängen als von den vorhandenen Rohdaten.
• • • •
Mit einem Stern* versehene Begriffe werden im Glossar erklärt.
Generelle Probleme phylogenetischer Rekonstruktionen
Ein widerspruchsfreier Stammbaum aller Lebewesen – seit der Zeit Darwins ist das der Traum aller Befürworter der Evolutionslehre. Im Idealfall sollten Analysen verschiedener Merkmale der untersuchten Organismen – von der Anatomie bis zur Genetik – zu denselben Ähnlichkeitsbeziehungen führen und so eine stimmige Stammbaumrekonstruktion ermöglichen. Angesichts vieler Schwierigkeiten mit unerwarteten Konvergenzen* im anatomischen Bereich und „schockierenden Homologien“ auf der genetischen Ebene rückt dieses Ziel schon bei höheren Lebewesen in weite Ferne (Braun 2012). Doch das eigentlich noch größere Problem befindet sich an der Wurzel des erhofften Stammbaums. Die Wurzel des Stammbaums repräsentiert die „frühe Evolution“, wo sich die Aufspaltung in die drei Domänen des Lebens Archaea, Bakterien und Eukaryoten* abgespielt haben soll (Woese 1998, Abb. 1). Allerdings gibt es bezüglich der Abstammungsverhältnisse der drei Domänen der Lebewesen noch weit mehr Uneinigkeit als hinsichtlich der angenommenen weiteren Evolution der Eukaryoten (Hug et al. 2016). Übereinstimmung besteht aus evolutionstheoretischer Sicht im Wesentlichen darin, dass Prokaryoten* wegen ihrer vergleichsweise geringeren Komplexität und ihrem Vorkommen im unteren Bereich der Fossilabfolge die ältesten Lebensformen sein sollen. Inzwischen mehren sich aber Stimmen, die die Eukaryoten näher zu den Archaeen rücken, da sie angeblich von diesen abstammen (Doolittle 2020).
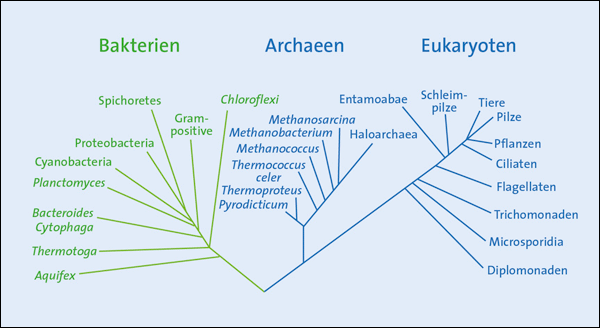
Abb. 1: Wurzel des hypothetischen Stammbaums aller Lebewesen nach Carl Woese (1998). Phylogenetische Analyse von Xavier et al.
Die größten Unsicherheiten des evolutionären Stammbaums des Lebens befinden sich an dessen Wurzel.
Für die Schwierigkeiten bei Rekonstruktionsversuchen der „frühen Evolution“ wird oft der Mangel an überlieferten Fossilien von frühen Mikroorganismen verantwortlich gemacht. Daher ist die phylogenetische* Analyse durch Vergleiche homologer* Gensequenzen die inzwischen fest etablierte Methode, um sich dieser Fragestellung anzunehmen. Wie auch bei anderen Argumenten auf der Basis von Vergleichen verschiedener Arten oder Organismengruppen beruht die phylogenetische Analyse aber auf der Voraussetzung, dass sich eine Makroevolution abgespielt hat. Folglich werden Unterschiede in Gensequenzen, die als homolog gelten, auf angehäufte Mutationen zurückgeführt. Gemäß dieser Denkweise gilt: je größer der Unterschied in einer homologen Gensequenz zweier Organismen, desto größer ihre stammesgeschichtliche Distanz.
Diese Vorgehensweise zur Rekonstruktion der hypothetischen Abstammungsverhältnisse ist jedoch im Fall von Mikroorganismen aufgrund des horizontalen Gentransfers (HGT)* erschwert. Das liegt daran, dass der HGT ein vom klassischen vertikalen Gentransfer (VGT)* unabhängiges Übertragen und Integrieren genetischer Information innerhalb derselben Generation und zwischen verschiedenen Bakterienarten ermöglicht. Dies ist aber ein großer Störfaktor für das Aufstellen von Hypothesen zu Abstammungsverhältnissen. Unter anderem aus diesem Grund bezeichnete der Biochemiker Frank Harold die angenommene frühe Evolution als „dunklen Bereich“. Zudem stellen Phänomene wie Rückmutationen* oder Mutations-Hotspots* weitere grundsätzliche Probleme für die Ableitung von Abstammungsverhältnissen aus dem Vergleich von Gensequenzen dar.
Kürzlich wurde in einigen Veröffentlichungen über den Versuch berichtet, den mutmaßlichen gemeinsamen Vorfahren aller Bakterien (last bacterial common ancestor, LBCA) zu charakterisieren, um zumindest in diesen Teil des dunklen Bereichs Licht zu bringen. Die Gruppe um Joana Xavier von der Universität Düsseldorf wählte für ihre Analyse zunächst aus in einer Datenbank (GenBank, NCBI) vorliegenden 5443 bakteriellen Genomen 1089 Exemplare aus, die anaeroben* Bakterien zugeordnet werden (Xavier et al. 2021). Auf diese Weise beschränkten sie a priori die Suche nach dem Mikroorganismus, der dem hypothetischen Vorfahren aller Bakterien am nächsten kommt, ausschließlich auf sauerstofffrei lebende Bakterien. Sie begründeten ihre Herangehensweise mit der verbreiteten Annahme einer frühen sauerstofffreien Atmosphäre: „[…] das Auftreten des Sauerstoffs veränderte weder die Beschaffenheit der grundlegenden [molekularen] Bausteine noch hat es ihre Biosynthesewege neu erschaffen. […] die Grundlagen der Biochemie, des Stoffwechsels und der Physiologie wurden zu einer Zeit erfunden, als die Erdatmosphäre anoxisch* war.“ (Hervorhebung hinzugefügt)
Die Autoren verwiesen in diesem Zusammenhang darauf, dass nur solche Genome ausgewählt wurden, in denen nachweislich die Information für die Synthese von Sauerstoff-Reduktasen* fehlte. Aus diesen 1089 Genomen identifizierten sie 146 Genfamilien, die für Stoffwechsel-relevante Proteine codieren. Auf diese Weise hofften sie, den Kern der bakteriellen Stoffwechsel-Netzwerke zu identifizieren und so auf den gemeinsamen Vorfahren schließen zu können. Bisher galt es als wenig aussichtsreich, auf der Grundlage von Genen für Stoffwechsel-Proteine Abstammungen zu rekonstruieren, da diese Gene unter Bakterien häufiger durch HGT ausgetauscht werden als solche, die für die Informationsverarbeitung zuständig sind. Xavier et al. sind sich dieser Schwierigkeit durchaus bewusst. Ihre Gewissheit, dass eine Rekonstruktion des LBCA dennoch gelingen muss, beruht auf einer nicht weiter belegten Annahme: „Metabolische Netzwerke und metabolische Enzyme bezeugen unzweifelhaft den evolutiven Prozess, doch fehlt es bisher an Methoden, die evolutionäre Information zu nutzen.“
Um ihren Ansatz zu plausibilisieren, greifen Xavier et al. zu einigen Hilfshypothesen. Sie gehen davon aus, dass der HGT erst nach der Anreicherung der zuvor anoxischen Atmosphäre mit Sauerstoff (great oxygenation event, GOE) einsetzte, sodass davor nur der VGT, also Weitergabe von Genen durch die Abstammungsabfolge, stattgefunden haben soll. Damit wäre das Verwischen vermutlicher Nachweise der gemeinsamen Abstammung durch den HGT zumindest in der Zeit vor dem GOE nicht mehr gegeben. Zur Stützung dieser These verweisen Xavier et al. auf eine andere Veröffentlichung, in der ebenfalls aus vergleichenden Erbgut-Analysen an Bakterien Schlussfolgerungen gezogen werden (Soo et al. 2017). Dort wird aber tatsächlich an keiner Stelle behauptet, dass der HGT erst mit dem GOE eingesetzt hätte. Darüber hinaus beruht der GOE auf einer unsicheren Datenbasis und ist deshalb keineswegs unumstritten (Schmidtgall 2021).
Die Annahmen der Abwesenheit des HGT bei frühen Organismen und des Vorhandenseins multifunktionaler Enzyme sind unbegründet und unplausibel.
Eine weitere Annahme, die Xavier et al. wiederholt anführen, ist das frühe Vorhandensein multifunktionaler Enzyme, also von Enzymen, die mehrere verschiedene Aufgaben erfüllen können. Sie beabsichtigen damit, die anfänglich kleine Zahl an metabolisch wirksamen Proteinen und dafür erforderlichen Genen im LBCA zu erklären: „Die Rekonstruktion des Stoffwechsels von LBCA weist auf das Vorhandensein mehrerer multifunktionaler Enzyme hin, sodass die Anzahl der für die Lebensfähigkeit erforderlichen Gene reduziert wird. Es handelt sich dabei […] möglicherweise um eine generelle Strategie bei frühesten Prokaryoten.“ Angesichts des sehr anspruchsvollen Aufbaus multifunktionaler Enzyme wird also ohne weitere Begründung eine sehr hohe Komplexität bereits zu einem sehr frühen Zeitpunkt der Evolution angenommen. Dies ist in Anbetracht der außerordentlich geringen Wahrscheinlichkeit der Entstehung selbst kleiner funktionaler Enzyme eine völlig unhaltbare Grundvoraussetzung (Thorvaldsen & Hössjer 2020). Auch die Bezugnahme auf den Nachweis einer funktionalen Vielseitigkeit (allerdings mit geringer Effizienz) bestimmter Enzyme (Khersonsky et al. 2006) ist nicht hilfreich, da in vielen Bereichen des Stoffwechsels eine sehr hohe Effizienz und Spezifität der Enzyme für das Überleben des jeweiligen Organismus unverzichtbar sind – besonders im Bereich des Energiehaushalts (Orgel 2007).
Als Resultat der phylogenetischen Analyse von Xavier et al. ergab sich, dass die den Firmicuten* zugehörigen Clostridien von ihrer Beschaffenheit her dem LBCA am nächsten stehen. Dementsprechend heißt es in der Arbeit, dass der gemeinsame Vorfahr aller Bakterien bereits folgende biomolekulare Einrichtungen aufwies: eine Zellwand, einen Zellteilungsapparat, die Erzeugung von Acetyl-CoA und Pyruvat, die Synthese von Zuckern (Gluconeogenese), nahezu alle Komponenten der Proteinbiosynthese (inklusive ihrer eigenen Biosynthese) sowie einige weitere Stoffwechselvorgänge. Auf die nahe liegende Frage, wie ein solch komplexer Organismus entstanden sein könnte, wird jedoch in keiner Weise eingegangen.
Phylogenetische Analyse von Williams et al.
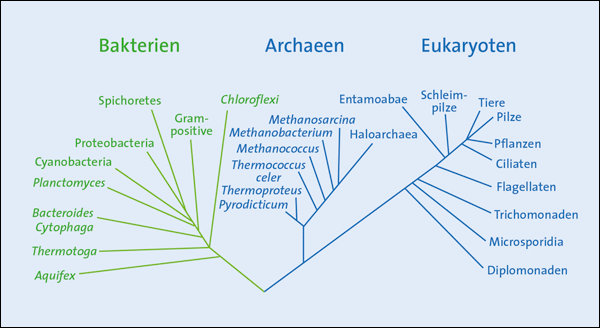
Abb. 2: Graphische Zusammenfassung der Arbeit von Williams et al (2021). Es werden darin die wichtigsten Ergebnisse der Arbeit zusammenfasst: Der gemeinsame Vorfahr aller Bakterien wird zwischen den zwei großen Stämmen Gracilicutes und Terrabacteria angesiedelt, während die Einordnung der Fusobacteria in den Stammbaum nicht gelang (links oben). Der Anteil der vom horizontalen Gentransfer betroffenen Gene wurde auf 34% geschätzt (rechts oben). Bezüglich der Ausstattung besaß der hier skizzierte LBCA folgende Eigenschaften: Es handelte sich um ein stabförmiges Bakterium mit einer Doppelmembran, äußerer Zellwand, Fortbewegungsfähigkeit (Flagellae) und Chemotaxis (unten).
Eine weitere Arbeit zur gleichen Fragestellung wurde von der Gruppe um Tom A. Williams von der Universität Bristol veröffentlicht (Coleman et al. 2021, Abb. 2). Sie erstellten einen Stammbaum durch Abstimmung verschiedener Stammbäume auf der Grundlage von 11.272 als homolog geltenden Genfamilien aus 265 bakteriellen Genomen (Genome taxonomy database, GTDB). Es wurde also für jedes zu einem Protein gehörige Gen ein Stammbaum der Bakterien erstellt. Anschließend wurden die 11.272 resultierten Stammbäume zu einem einzigen harmonisiert. Dabei wurden statistische Unsicherheiten der genetischen Stammbäume und eine Abschätzung der Raten von Gen-Duplikationen, -Verlusten und -Übertragungen einbezogen. Im Unterschied zu Xavier et al. setzten sie allerdings nicht a priori voraus, dass der LBCA unter den Anaerobiern anzusiedeln ist. Sie sahen es aber als erforderliche Prämisse an, dass der gemeinsame Vorfahr aller Lebewesen (last universal common ancestor, LUCA) zwischen Bakterien und Archaea liegen muss. Dabei betonen auch sie, dass „die verfügbaren Indizien beschränkt und schwierig zu interpretieren sind.“
Williams et al. sehen im HGT ebenfalls ein ernsthaftes Hindernis für die Rekonstruktion von Abstammungsverhältnissen: „Ein weitreichender HGT stellt eine existentielle Herausforderung für die [stammesgeschichtliche] Verkettung dar, weil er die Anzahl der Gene, die sich auf einem einzelnen zugrunde liegenden Stammbaum entwickeln, in hohem Maße verschleiert.“ Sie schätzen, dass bakterielle Genome im Mittel zu etwa einem Drittel vom HGT betroffen sind – ein sehr hoher Anteil und damit ein großer Unsicherheitsfaktor hinsichtlich der Rekonstruktion von theoretischen Abstammungsverhältnissen.
Dennoch sind sie sicher, den vertikalen Anteil der Vererbung identifizieren zu können. Auf dieser Information aufbauend skizzieren die Autoren um Williams in ihrer Veröffentlichung ein sehr detailreiches Porträt des LBCA: „Wir nehmen an, dass der letzte gemeinsame bakterielle Vorfahr eine freilebende, begeißelte, stäbchenförmige Zelle war, die eine Doppelmembran mit einer Lipopolysaccharid-Außenschicht, ein Typ-III-CRISPR-Cas-System [Immunsystem], Typ-IV-Pili und die Fähigkeit zur Wahrnehmung und Reaktion durch Chemotaxis aufweist.“ Weitere Komponenten wie der Zellteilungsapparat, Flagellae, die Proteinbiosynthese, der Zitratzyklus und der Pentosephosphatweg wurden neben einigen weiteren auch dazu gezählt. Die Beschreibung ist damit auffällig nah an der typischen Ausstattung vieler heutiger Mikroorganismen. Unwillkürlich stellt sich die Frage, ob in der angenommenen Zeit nach dem LBCA von immerhin ca. 3 Milliarden (radiometrischen) Jahren überhaupt noch signifikante Veränderungen der Mikroorganismen durch Evolution stattgefunden haben können. Die Konsequenz wäre demnach, dass die eigentlich interessantesten „evolutiven Vorgänge“ in der dunklen Zeit vor dem LBCA liegen müssten.
Verhaltene Kritik an der Arbeit von Williams äußert Katz in einem kurzen Kommentar der Zeitschrift Science: „Obwohl Coleman et al. den vertikalen Teil des bakteriellen Lebensbaums akkurat erfasst haben mögen, fehlt dem Baum, den sie verwurzeln, die Geschichte der biologischen Innovationen und ökologischen Anpassungen, die von LGTs abgeleitet sind“ (Hervorhebung hinzugefügt; LGT, lateraler Gentransfer, ist dasselbe wie HGT). Dabei ließ Katz unerwähnt, dass auch die Darstellung des „vertikalen Teils“ eines hypothetischen Stammbaums (d. h. nach Abzug der vermutlich horizontal übertragenen Gene) keinerlei Information darüber enthält, durch welche Vorgänge es zu den entscheidenden Innovationen im Stoffwechsel und der molekularen Ausstattung der Mikroorganismen gekommen sein könnte.
Eine wesentliche Triebfeder der modernen Evolutionsforschung besteht darin, evolutionäre Stammbäume zu „retten“.
Abschließend fügte Katz dann noch hinzu: „Es wurde vorgeschlagen, dass man ‚zur Rettung der Bäume Organismen als mehr als die Summe ihrer Gene definieren und sich vorstellen könnte, dass die Abstammungslinien von Organismen eine Art emergente Realität haben‘.“ (Hervorhebungen hinzugefügt)
Offenbar besteht eine wesentliche Triebfeder der modernen Evolutionsforschung darin, die evolutionären Stammbäume zu retten und den Fokus auf der als unbezweifelbar hingestellten evolutionären Geschichte zu behalten auch angesichts immenser Schwierigkeiten, sie mit den Daten in Einklang zu bringen. Zur Not kann man sich auch mit der Vorstellung einer „emergenten Realität“ behelfen – was auch immer damit gemeint sein soll.
Glossar
anaerob: unter Ausschluss von Sauerstoff lebend. anoxisch: frei von Sauerstoff (in Bezug auf die Atmosphäre). Eukaryoten: Lebewesen, deren Zellen über einen Zellkern verfügen. Firmicuten: eine Klassifikationsebene von Bakterien, die den Rang eines Phylums (Stamm) hat. Die meisten Gram-positiven Bakterien gehören zu diesem Phylum. homolog: als „homolog“ werden in der Biologie gestaltlich ähnliche Merkmale verschiedener Arten bezeichnet, wobei angenommen wird, dass die Ähnlichkeit durch gemeinsame Abstammung zustande gekommen ist. horizontaler Gentransfer: Bakterien können Erbgut-Moleküle untereinander über ihre Pili (aus Proteinen bestehende feine Röhren) übertragen. Anschließend können diese ins Erbgut integriert werden. Dieser Gentransfer kann auch unter verschiedenen Arten von Bakterien stattfinden und ist in keiner Weise an die Abstammung gebunden. Konvergenz: Zwei- bis mehrfache Entstehung weitgehend baugleicher Merkmale oder identischer DNA-Sequenzen bei nicht abstammungsverwandten Formen. Mutations-Hotspot: Das Mutationsgeschehen im Erbgut von Lebewesen ist nicht gleichmäßig verteilt. Stellen, an denen deutlich öfter Mutationen eintreten, werden als „Mutations-Hotspots“ bezeichnet. phylogenetisch: „Phylogenese“ ist ein zusammengesetztes Wort aus „Phylum“ (Stamm) und „Genese“ (Entstehung). „Phylogenetisch“ bedeutet also: Mit Bezug zur Entstehung der Tierstämme. Prokaryoten: Einzeller, die keinen Zellkern aufweisen. Rückmutation: Mutationen sind in vielen Fällen umkehrbar. Durch Rückmutationen werden also erfolgte Mutationen wieder rückgängig gemacht. Sauerstoff-Reduktasen: Enzyme, die Elektronen von organischen Molekülen auf Sauerstoff übertragen und dadurch das Sauerstoff-Molekül in andere chemische Verbindungen einbauen (z. B. Wasserstoff-Peroxid, Wasser). vertikaler Gentransfer: Übertragung der genetischen Information von der Elterngeneration auf die Nachkommen, also durch Abstammung.
Fazit
Zusammenfassend kann gesagt werden, dass die Versuche, Stammbäume der bakteriellen Evolution aufzustellen und einen gemeinsamen Vorfahren aller Bakterien zu charakterisieren, sowohl hinsichtlich ihrer Grundannahmen als auch der daraus abgeleiteten Methoden alles andere als solide begründet sind. Wie die Arbeiten von Xavier und Williams zeigen, ist die a priori Einordnung des LBCA (bzw. des LUCA) im evolutionären Stammbaum entscheidend für das Ergebnis. Die Voraussetzung, dass es sich um anaerobe Organismen gehandelt haben muss, beruht auf unsicheren Dateninterpretationen im Zusammenhang mit der vermuteten Anreicherung einer zuvor anoxischen Atmosphäre mit Sauerstoff (GOE). Auch die Annahme von Xavier, dass es vor dem GOE keinen HGT gab, ist offenbar eine bloße Behauptung, die nicht einmal in der zitierten Quelle zu finden ist. Eine entscheidende methodische Schwäche der beiden Arbeiten ist die Unsicherheit bezüglich der Frage, welche Gene vom HGT betroffen sind und welche nicht. Interessanterweise stellen sowohl Xavier als auch Williams den LBCA als vollständiges Bakterium dar, sodass die wesentlichen biologischen Innovationen sich davor hätten ereignen müssen. Über das Zustandekommen dieser Innovationen geben die Veröffentlichungen jedoch keinerlei Auskunft.
Die hier beschriebenen Annahmen und Methoden scheinen notwendig zu sein, um die immense Vielfalt und die Flickenteppich-artig verteilten Merkmalsmuster der Mikroorganismen in ein evolutionstheoretisches Konzept fügen zu können. Alternativ können die Bakterien und Archaea mit ihren vielseitigen metabolischen Eigenschaften als intelligent geschaffene Organismen verstanden werden, deren Vielfalt notwendig ist für die Vitalität und Stabilität vieler verschiedener Ökosysteme. Eine intelligente Schöpfung ist überdies die einzige plausible Erklärung für die offenbar sehr hohe minimale Komplexität von Mikroorganismen.
Warten auf einen neuen Einstein. Stud. Integr. J. 19, 12–19.
A rooted phylogeny resolves early bacterial evolution. Science 372, eabe0511.
Evolution: Two domains of life or three? Curr. Biol. 30, R159–R179.
In search of cell history: the evolution of life’s building blocks. The University of Chicago press.
A new view of the tree of life. Nat. Microbiol. 1, 16048.
Illuminating first bacteria. Science 372, 574–575.
Enzyme promiscuity: evolutionary and mechanistic aspects. Curr. Opin. Chem. Biol. 10, 498–508.
The implausibility of metabolic cycles on the prebiotic earth. PloS Biology 6, e18.
Die „Sauerstoffkatastrophe“. Stud. Integr. J. 28, 13–21.
On the origins of oxygenic photosynthesis and aerobic respiration in Cyanobacteria, Science 355, 1436–1444.
Using statistical methods to model the fine-tuning of molecular machines and systems. J. Theor. Biol. 501, 110352.
Default taxonomy: Ernst Mayr’s view of the microbial world. Proc. Natl. Acad. Sci., doi:10.1073/pnas.95.19.11043.
The metabolic network of the last bacterial common ancestor. Nat. Comm. 4, doi: 10.1038/s42003-021-01918-4.
Themen | Kurzbeiträge | Streiflichter
Studiengemeinschaft WORT und WISSEN e.V.
Letzte Änderung: 11/24/21
Webmaster