Themen • Kurzbeiträge • Streiflichter
„Wenn ENCODE richtig liegt, dann ist Evolution falsch“
Wie ENCODE unser Verständnis des Erbguts veränderte
von Peter Borger
Studium Integrale Journal
28. Jahrgang / Heft 1 - April 2021
Seite 30 - 37
Zusammenfassung: Die Sequenz der 3 Milliarden DNA-Buchstaben des gesamten menschlichen Erbguts wurde 2001 nach mehr als zehn Jahren intensiver Arbeit veröffentlicht. Aber welche Informationen und damit Funktionen beinhalten diese Sequenzen? Die Entschlüsselung der funktionellen Elemente des menschlichen Genoms durch ENCODE hat die Sichtweise auf das menschliche Genom enorm verändert. Es erweist sich als ein fast vollständig funktionsfähiger superdynamischer Informationsverarbeitungscomputer und es gibt kaum noch Abschnitte, die man als „Junk-DNA“ bezeichnen könnte.
• • • • • • •
Die DNA und das Erbgut
Als Beginn der Geschichte um das Erbgut kann man das Jahr 1869 betrachten, als der Schweizer Biochemiker Friedrich Miescher das Erbmaterial isolierte. Diesem Stoff, für den Miescher den Begriff „Nuklein“ prägte, wurde anfangs nur wenig Beachtung geschenkt. Die überwiegende Mehrheit der Wissenschaftler einschließlich Miescher war damals davon überzeugt, dass die komplexen Proteine die Träger der genetischen Information sein müssten. Man glaubte, dass die Vielfalt der Erbinformation ähnlich wie in unserem 26-teiligen Alphabet in der Abfolge der 20 Aminosäuren zu finden sei. Die neuentdeckte Kernsubstanz bestand aus nur vier Nukleotiden – viel zu wenig, so glaubte man, um die enorme Menge an genetischer Information zu speichern.
In den ersten vier Jahrzehnten des 20. Jahrhunderts gab es ein allmählich zunehmendes Interesse am „Nuklein“. Dem deutschen Arzt und Biochemiker Albrecht Kossel war es gegen Ende des 19. Jahrhunderts gelungen, die vier Nukleobasen der DNA (Cytosin, Thymin, Adenin und Guanin) zu isolieren, wofür er 1910 den Nobelpreis erhielt. Wichtige Beiträge, die dazu führten, dass die Bedeutung der DNA als Träger der Erbinformation zunehmend erkannt worden ist, leisteten auch Oswald Avery durch seine Arbeit an Bakterien und die Chemiker Linus Pauling und Erwin Chargaff. Vorträge des österreichischen Physikers Edwin Schrödinger am Trinity College in Dublin, die 1944 unter dem Titel „What is Life?“ veröffentlicht wurden (Schrödinger 1944), zeigten eine weitreichende Wirkungsgeschichte. Schrödinger verstand, dass die Ordnung in den biologischen Organismen nicht auf das statistische Verhalten von Molekülen zurückzuführen ist, sondern auf codierte Anweisungen. In diesem Buch sagte er voraus, wie die Grundlage des Lebens beschaffen ist: Es wird kein periodischer Kristall sein, denn diese sind langweilig. Er vermutete, dass das Erbgut von Lebewesen aus „aperiodischen Kristallen“ mit einem Mikrocode (d. h. Information) besteht. Mutig behauptete er, dass sowohl die Anweisungen zum Aufbau des Organismus als auch der Mechanismus zu dessen Erzeugung in den Chromosomen enthalten sind. Eine sowohl brillante als auch visionäre Idee.
Im gleichen Jahr wurde experimentell belegt, dass die DNA tatsächlich die genetische Information trägt (Avery et al. 1944). Sie besteht aus vier Arten von kleineren Molekülen, die als „Nukleotide“ bezeichnet werden: Adenosin (A), Thymidin (T), Cytidin (C) und Guanosin (G). Knapp eine Dekade später, im Jahr 1953, beschrieben James D. Watson und Francis Crick mit Hilfe der Röntgenstrukturanalysen von Rosalind Franklin (die Crick und Watson heimlich eingesehen hatten!) die chemische Struktur des DNA-Moleküls. Die dreidimensionale Struktur der Doppelhelix, die sie beschrieben, machte verständlich, wie die Eigenschaften der Elterngeneration auf die Nachkommen übertragen werden. Die Reihenfolge der Basen liefert die Anweisungen für den Zusammenbau der wesentlichen Bausteine des Lebens.
Mit einem Stern* versehene Begriffe werden im Glossar erklärt.
Seit 1961 wird das Genom nicht nur als eine Sammlung von Bauplänen betrachtet, sondern als eine Gesamtheit von aufeinander abgestimmten Programmen, die die Synthese von Proteinen koordiniert und kontrolliert (Jacob & Monod 1961). Im 21. Jahrhundert stellen wir fest, dass eine lebende Zelle einem Datenprozessor vergleichbar ist und analog zu einem Computer funktioniert. Der Computer auf Ihrem Schreibtisch hat eine Tastatur, die Eingaben von außen ermöglicht (Input). Im Inneren gibt es eine Festplatte mit intelligent konzipierten Programmen zum Speichern und Verarbeiten des Inputs, und es gibt einen Bildschirm – oder ein anderes Gerät – für die Darstellung des Outputs als Resultat der Informationsverarbeitung. In Analogie dazu hat die lebende Zelle Sensoren, um Reize aus der Umwelt wahrzunehmen (Input), das Erbgut-Molekül DNA als System zur Datenspeicherung und -verarbeitung mit der RNA* als mobilem Zwischenspeicher, sowie einen biologischen Output, meist in Form von Stoffwechselvorgängen und letztlich biologischen Strukturen und Verhaltensweisen. Bei bestimmten Eingaben der zellulären Umgebung passt die Zelle die genetische Aktivität an.
Zusammenfassend kann also gesagt werden: Eine lebende Zelle ist ein auf DNA basierender RNA-Computer mit Protein-Output. Der Begriff „Genom“ (für Erbgut) wurde erstmals 1920 von Hans Winkler geprägt, einem Professor für Botanik an der Universität Hamburg. Er schlug vor, den haploiden* Chromosomensatz, der zusammen mit dem dazugehörigen Protoplasma die materielle Grundlage der Zelle bildet, als „Genom“ zu bezeichnen (Winkler 1920). Das Genom umfasst die gesamte DNA der Zelle, einschließlich der DNA in Mitochondrien und Chloroplasten (bei Pflanzen). Grundsätzlich umfasst das Genom alle DNA-Sequenzen eines Organismus.1
Kompakt
Nachdem im Jahr 2001 das menschliche Genom* sequenziert und der Öffentlichkeit präsentiert worden war, vermutete man, dass die Lösung aller genetischen Grundfragen und Erbkrankheiten nur noch eine Frage der Zeit sein würde. Wie liest man jedoch eine Anleitung, ohne die Sprache zu verstehen, in der sie geschrieben ist? Um die Bedeutung aller 3 Milliarden DNA-Buchstaben (Nukleotide) zu erforschen, wurde ein internationales Konsortium von Genomforschern gegründet: ENCODE. Seit 2003 arbeiten die Forscher an der Entschlüsselung der funktionalen Elemente des menschlichen Genoms. An ENCODE beteiligte Forschergruppen veröffentlichen in einer Reihe von Publikationen, dass entgegen der lange geglaubten Annahme, das menschliche Genom sei voll von „Junk-DNA“ (DNA-Müll), mindestens 80% des Genoms eine Funktion haben. Fast jede DNA-Sequenz im Genom führt irgendeine Funktion aus und/oder wird in ein RNA-Transkript* umgeschrieben. Obwohl diese Erkenntnisse unter Evolutionsbiologen heftig umstritten sind, da ein fast vollständig funktionsfähiges Genom den Evolutionsprozess grundlegend in Frage stellt, lieferte ENCODE im Jahr 2020 weitere Beweise dafür, dass das Genom Millionen von regulatorischen Sequenzen und genetischen Schaltern enthält, um die biologischen Leistungen des Organismus zu ermöglichen oder zu optimieren. Das Genom, das von ENCODE beschrieben wird, lässt sich am besten als „RNA-Computer mit Protein-Output“ verstehen. Die DNA-Information erweist sich als überlappend, mehrschichtig und mehrdimensional und darüber hinaus in hohem Maße dynamisch. Sie wird sowohl vorwärts als auch rückwärts gelesen und es gibt kaum noch Abschnitte, die man als „Junk-DNA“ bezeichnen könnte. Die neuen Daten von ENCODE stellen einen ungerichteten Evolutionsprozess, wie es derzeit gelehrt wird, abermals in Frage. Oder, wie der Evolutionsbiologe Dan Graur es 2012 formulierte: „Wenn ENCODE richtig liegt, ist die Evolution falsch.“
HUGO und ENCODE
Das Genom des Menschen besteht aus der in 23 Chromosomen verteilten DNA und der mitochondrialen (mt) DNA.2 Insgesamt beträgt die Summe der DNA-Buchstaben (Nukleotide) etwa 3 Milliarden, die aneinandergereiht im Zellkern eine molekulare Kette von etwa 1 Meter Länge ergeben. Dies ist unser genetischer Bauplan, darin ist die genetische Information in sehr dichter Form abgebildet. Von den 1980er- bis zu den 1990er-Jahren wurden große Fortschritte bei der Entschlüsselung genomischer Information erzielt. Aber es war eine langsame und aufwendige Arbeit, die von unabhängigen und selbständig arbeitenden Wissenschaftlern durchgeführt wurde. Erst der Start des internationalen HUGO-Projekts (HUGO ist das Kürzel für Human Genome Organisation) im Jahr 1988 ermöglichte einen systematischen Ansatz zur Entschlüsselung der kompletten menschlichen DNA-Sequenz, einschließlich der Anordnung unserer Gene und ihrer Verteilung auf den Chromosomen. Die Fertigstellung eines „Arbeitsentwurfs“ des menschlichen Genoms wurde am 26. Juni 2000 auf einer gemeinsamen US/UK-Pressekonferenz von Präsident Clinton und Premierminister Blair bekannt gegeben.
Die Ergebnisse aus HUGO zeigten, dass das Genom des Menschen überraschenderweise nur etwa 20.000-25.000 Protein-Gene umfasst.
Die Ergebnisse aus HUGO zeigten, dass das Genom des Menschen überraschenderweise nur etwa 20.000-25.000 Protein-Gene* umfasst (aktuelle Schätzungen gehen vom unteren Ende dieses Bereichs aus, also etwa 20.000)3 (Nature Definitions 2021). Die Entschlüsselung des Genoms stellte nichts Geringeres in Aussicht als die Offenlegung des Bauplans der menschlichen Biologie. In euphorischer Stimmung berichteten viele Medien, dass, weil wir nun das Genom kennen, genetische Krankheiten bald der Vergangenheit angehören würden. Zehn Jahre danach hieß es in der Zeitschrift Nature kleinlaut: „Wir haben uns darin getäuscht, dass das Genom eine transparente Bauanleitung sein würde, aber das war es nicht“ (Hayden 2010). Es liegt eine gewisse Naivität in der Vorstellung, dass es ausreicht, eine sehr große Menge an Sequenzen zu analysieren, um zu verstehen, was in einer Zelle vor sich geht. Die DNA enthält lineare Information, die für dreidimensionale Proteine codiert, deren Funktion und Menge sich mit der Zeit ändern. Hinzu kommt, dass Gene nie alleine, sondern in kooperierenden Netzwerken funktionieren. Außerdem ist die Regulation der Genexpression* beim Menschen (und anderen eukaryotischen Zellen) völlig anders als bei Bakterien. Letztere ist mehr oder weniger entschlüsselt und basiert auf linearen Funktionseinheiten (Operons), genetischen Programmen, die kolinear auf dem einzelnen bakteriellen Chromosom zu finden sind, in denen Gene gleichzeitig oder nacheinander aktiviert werden. In Organismen mit mehr als einem Chromosom müssen genetische Plattformen (vergleichbar mit bakteriellen Operons) konstruiert werden, um die richtigen Gene, die sich oft auf verschiedenen Chromosomen befinden, für die Transkriptionsmaschinerie zugänglich zu machen. Nur im richtigen raumzeitlichen Zusammenhang können mehrere biologische Funktionen zeitgleich abgerufen werden. Es erfordert eine Menge Rechenleistung (Auslesen von Sequenzen), um die 1-dimensionale DNA-Information im richten Moment und in den richtigen Zellen in 4-dimensionale (d. h. in Raum und Zeit) biologische Funktionen umzuwandeln.
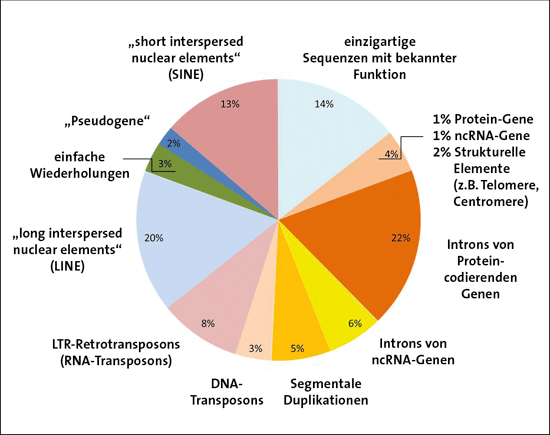
Abb. 1: Die verschiedenen funktionellen Teile des Genoms. Vor ENCODE wurden nur 4-5% des Genoms als biologisch-funktionelle Sequenzen angesehen. Bis zu 95% wurde als „junk DNA“ betrachtet. Nachdem die ersten beiden Phasen von ENCODE veröffentlicht wurden, wurde der funktionelle Anteil auf maximal 25% geschätzt (Graur 2017). Je tiefer Genomforscher in die DNA schauen, desto mehr Funktion entdecken sie. Die neueste Erkenntnis ist die Existenz von Regionen mit langen strukturellen DNA-Elementen, die für die Entstehung der räumlichen Konfiguration der Chromosomen verantwortlich sind.
Das Genom lässt sich am besten mit einer codierten Anleitung vergleichen, deren Bedeutung uns größtenteils noch unbekannt ist. Die Frage ist: Wie liest man eine Anleitung, ohne die Sprache zu verstehen, in der sie geschrieben ist? Zu der Zeit, als das menschliche Genom entschlüsselt wurde, war nur der Teil des Erbguts gut verstanden, der für Proteine codiert. Dieser Teil macht jedoch lediglich maximal 2 % des menschlichen Genoms aus (sog. „Protein-Code“). Man ging immer davon aus, dass diese 2 % und ungefähr weitere 3-5 %, die zur Expression des Protein-Codes benötigt werden („Schaltsequenzen“), das gesamte funktionale Genom darstellen. Die gängige Lehrmeinung war (und wird vereinzelt immer noch vertreten), dass etwa 90-95 % des Genoms ohne Funktion sind und daher als „Junk-DNA“ („Abfall-DNA“) betrachtet werden sollten, d. h. als funktionslose Überbleibsel des evolutionären Prozesses (siehe Kasten).
Abb. 1 zeigt die genetischen Elemente und Sequenzen, die im menschlichen Genom zu einem bestimmten Prozentsatz vorhanden sind. Proteincodierende Gene nehmen etwa 23 % des Genoms ein (1 % Exons*, 22 % Introns*). Gene für funktionelle nichtcodierende RNAs (ncRNA) nehmen weitere 7 % des Genoms ein (1 % Exons, 6 % Introns). Beim Abschreiben eines Gens (Transkription) werden jedoch nicht nur die Exons (der proteincodierende Teil des Gens) abgeschrieben, sondern auch die Introns. Wenn wir also ein Gen als eine DNA-Sequenz definieren, die transkribiert (in RNA umgeschrieben) wird, sind etwa 30 % des Genoms Gene. Das menschliche Genom enthält jedoch weit mehr als nur Gene. Etwa die Hälfte des Genoms besteht aus Transposons* und transposonähnlichen genetischen Elementen. Man unterscheidet DNA-Transposons (3%), RNA-Transposons (auch: endogene Retroviren, ERV; 8%), LINEs* (20%) und SINEs* (13%). Sie werden evolutionstheoretisch als Überbleibsel alter Einfügungen durch Viren interpretiert, insbesondere weil RNA-Transposons („endogene Retroviren“) einen ähnlichen Aufbau haben wie bestimmte RNA-Viren4. Obwohl sie alle mit dem bekannten vierbuchstabigen DNA-Alphabet (A, C, G und T) geschrieben sind, ist die biologische Bedeutung der meisten dieser DNA-Sequenzen immer noch ungeklärt. Sind sie funktionslos? Oder haben sie biologische, biochemische oder strukturelle Funktionen? Die Aktivität und Expression* von Genen zum richtigen Zeitpunkt und im richtigen Gewebe muss eng und präzise kontrolliert werden. Zu diesem Zweck gibt es eine Vielzahl an DNA-Elementen wie Promotoren*, transkriptionelle regulatorische Sequenzen und Regionen, die die dreidimensionale Anordnung des genetischen Materials kontrollieren, damit Genprogramme abgerufen oder geschlossen werden können. An dieser Stelle setzt das ENCODE-Projekt an (Abb. 2). Das primäre Ziel des ENCODE-Projekts ist es, eine umfassende Karte der funktionellen Elemente des Humangenoms zu erstellen.
Das Bild kann online nicht zur Verfügung gestellt werden.
Abb. 2: ENCODE ist ein großes internationales Konsortium von mehreren hundert Genomforschern, das vom National Human Genome Research Institute (NHGRI) im September 2003 ins Leben gerufen wurde, um alle funktionellen Elemente im menschlichen Genom zu identifizieren. (Quelle: https://www.nature.com/collections/aghcdefffg/)
ENCODE wurde vom US National Human Genome Research Institute (NHGRI) im September 2003 ins Leben gerufen. Ähnlich wie bei HUGO steht auch hinter dem ENCODE-Projekt ein großes internationales Konsortium von mehreren hundert Genomforschern. Das Ziel von ENCODE stellt den nächsten logischen Schritt aufbauend auf HUGO dar: die Identifikation aller funktionellen Elemente im menschlichen Genom. Der Begriff selbst, ENCODE, ist eine Abkürzung und steht für „ENCyclopedia of DNA Elements“ (Enzyklopädie der DNA-Elemente). Dieses Kürzel informiert bereits über das angestrebte Ziel: Alle funktionellen Elemente im menschlichen Genom sollten bestimmt (annotiert*) werden. Die Wissenschaftler wollten nicht nur alle Gene und die Schalter, die die Gene an- und ausschalten, lokalisieren, sondern auch herausfinden, wie viele RNA-Transkripte existieren und was sich sonst noch alles im Genom mit biochemischen oder genetischen Funktionen zeigt. Die an ENCODE beteiligten Wissenschaftler entwickelten neue Technologien der Genkartierung*, um biochemische Aktivitäten genetischer Regionen und damit Kandidaten für regulatorische Elemente ausfindig zu machen. Die Identifikation der funktionellen Elemente im menschlichen Genom durch ENCODE erfolgt in vier aufeinander folgenden Phasen. Im Juli 2020 wurde die dritte Phase abgeschlossen, deren wichtigste Ergebnisse in der Zeitschrift Nature veröffentlicht wurden.
Glossar
Annotieren: Die Annotation einer DNA-Sequenz beschreibt die genaue Lage von → Exons und → Introns sowie die repetitiven und funktionellen DNA-Elemente in diesen Sequenzen. Chromatin: Material, aus dem Chromosomen bestehen, ein Komplex aus DNA und speziellen Proteinen (→ Histonen). Chromatin kann dicht oder locker sein, je nachdem, wie es chemisch modifiziert wird. Enhancer: DNA-Sequenzen, die mit → Promotoren vergleichbar sind, aber im Gegensatz zu diesen nicht unmittelbar stromaufwärts der proteincodierenden Sequenz liegen, sondern in einem gewissen Abstand. Epigenetisch (Epigenetik): bezieht sich auf Faktoren, die vorübergehend die Aktivität eines Gens und damit die Entwicklung der Zelle bestimmen. Exon: Der Teil eines Gens, der genetische Information enthält und in mRNA abgeschrieben wird und nach einer weiteren Bearbeitung erhalten bleibt. Exons tragen die biologische Funktion eines Proteins oder einer nichtcodierenden RNA. Angrenzende Exons werden durch → Introns voneinander getrennt. Expression (Gene) / exprimieren: Ausprägen/Verwirklichen eines Gens im Erscheinungsbild (Phänotyp) eines Lebewesens. Genom: Das vollständige → haploide Erbgut eines Organismus. Haploid: Das Vorhandensein eines einfachen Chromosomensatzes. Histon: DNA-bindendes Protein. Histoncode: Spezifische → Histonmodifikationen, die die Übersetzung von genetischer Information kontrollieren. Histonmodifikationen: Chemische Veränderungen der → Histon-Proteine, die die Zugänglichkeit des → Chromatins für → Transkriptionsfaktoren beeinflussen. Immunpräzipitation: Labortechnologie, um mit Hilfe von Antikörpern Moleküle (z. B. Chromatinbausteine) zu binden und isolieren. Intron: Abschnitt der DNA innerhalb eines Gens, der benachbarte → Exons trennt. Introns werden → transkribiert (in prä-mRNA abgeschrieben), aber aus dieser herausgespleißt (ausgeschnitten), bevor sie in Proteine oder ncRNAs übersetzt wird. kartieren: Feststellen, welche Bereiche in RNA transkribiert werden und wo sie sich im Genom befinden. konserviert: In der Evolutionsbiologie werden DNA-Abschnitte, die bei vielen Organismen sehr ähnlich sind, als über lange Zeiträume unverändert und somit konserviert eingestuft. LINEs: Kürzel für „long interspersed nuclear elements”. Komplexe genetische Elemente, die sich mit Hilfe ihrer selbst-codierten Gene (Reverse Transkriptase und Integrase) im Genom vermehren und umlagern können. Sie werden evolutionstheoretisch als die Überbleibsel von uralten Vireninvasionen betrachtet („Junk DNA“). Methylierung: eine chemische Veränderung, bei der eine Methylgruppe durch Enzyme (DNA-Methyltransferasen) an die DNA übertragen wird. Promotor: eine Nukleotid-Sequenz, die die regulierte → Expression eines Gens ermöglicht. Protein-Gen: Gen, das für ein Protein codiert. Daneben gibt es RNA-Gene, die nur in RNA → transkribiert, aber nicht in Proteine → translatiert werden. RNA: ein einzelsträngiges, mit der DNA chemisch verwandtes Molekül. SINEs: Kürzel für „short interspersed nuclear elements”. Kleine genetische Elemente, die sich mit Hilfe von → LINEs im Genom vermehren und umlagern können. Sie werden als die Überbleibsel von degenerierten LINEs betrachtet („Junk DNA“). Transkription, transkribieren: Das Abschreiben von DNA in mRNA. Transkriptionsfaktor: (Komplex von) Proteinen (und manchmal auch RNA), der das Abschreiben (Transkription) eines Gens initiiert, indem er an DNA bindet und einen Ansatzpunkt für die RNA-Polymerase bildet. Transkriptom: Eine Momentaufnahme der gesamten RNA-Transkripte, die in einer Zelle oder im Gewebe vorhanden sind. Translation, translatieren: Übersetzung von mRNA in ein Protein. Transposon: genetisches Element (DNA-Sequenz), das sich im Genom umlagern kann.
Wie wird im Rahmen von ENCODE vorgegangen?
Phase 1 (2003–2007) war als eine Vorstudie gedacht, wobei man etwa ein Prozent des menschlichen Genoms untersuchte, um neu entwickelte Technologien zu bewerten. Die Hälfte von diesem einen Prozent befand sich in Genregionen von hoher funktioneller Relevanz, und die andere Hälfte wurde ausgewählt, um Erkenntnisse über die Nutzung des Genoms zu erhalten, insbesondere aus welchen Sequenzen Gene zusammengesetzt sind und wie sie transkribiert werden. Neue Technologien wurden eingesetzt, um festzustellen, wo Transkriptionsfaktoren* an diese DNA-Regionen binden. Außerdem wurden neue Methoden verwendet, um den Histoncode* zu identifizieren, der die Zugänglichkeit der DNA für Enzyme und andere regulatorische Makromoleküle durch Chromatinmodifikationen* reguliert (Abb. 3).
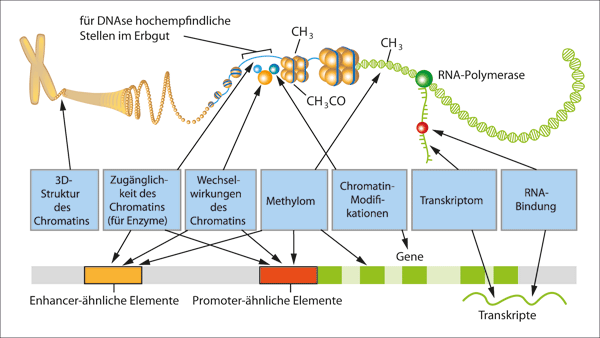
Abb. 3: Aufbau des Genoms in schematischer Darstellung. Chromosomen bestehen aus eng gewickeltem Chromatin, das aus der DNA-Kette besteht, die um Histon-Proteine gewickelt ist. Um genetische Programme mehr oder weniger zugänglich zu machen, werden DNA und Histone methyliert (eine -CH3-Gruppe angeheftet), während Histone auch acetyliert werden können (Anheftung von -CH3CO). ENCODE-Forscher untersuchen diese und andere Prozesse, die steuern, wie Gene an- und abgeschaltet werden (im Kasten unten erwähnt).
ENCODE fand heraus, dass für mehr als 80% des Genoms „biochemische Funktionen zugewiesen werden konnten, insbesondere außerhalb der gut untersuchten proteincodierenden Regionen.
In Phase 1 wurde herausgefunden, dass 93% der ausgewählten Teile des Genoms in RNA-Moleküle abgeschrieben werden (und nicht nur 1-2%, wie aufgrund des Protein-Codes erwartet wurde). Dabei gibt es eine ausgeprägte Mehrfachverwendung: Jeder beliebige Buchstabe des Genoms wird im Durchschnitt in sechs verschiedenen RNA-Transkripten verwendet. Fast das ganze Genom ist somit permanent damit beschäftigt, etwas zu tun (Birney et al. 2007). Da für die Transkription viel Energie und Koordination erforderlich ist, bedeutet dies, dass wahrscheinlich (fast) das gesamte Genom von der Zelle genutzt wird und es so etwas wie funktionslose „Junk-DNA“ gar nicht gibt. Die funktionelle Bedeutung dieser RNA-Transkripte, die man zusammen als Epitranskriptom bezeichnet, wurde in Phase 2 und 3 weiter analysiert.
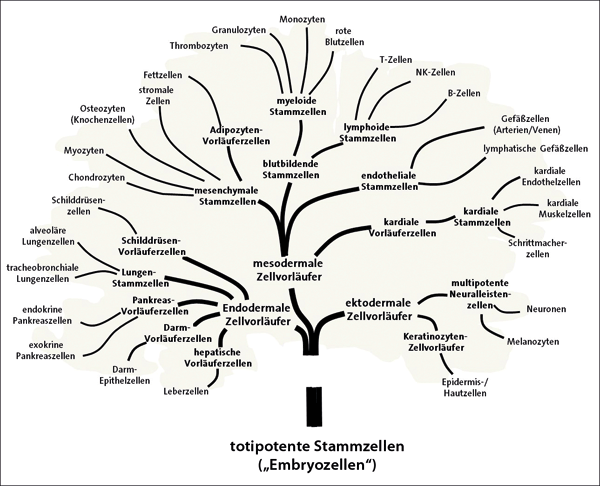
Abb. 4: Der Baum zeigt die Hauptwege im Prozess der zellulären Differenzierung (vereinfacht). Im Rahmen des ENCODE-Projekts wurden hunderte von unterschiedlich differenzierten Zellen des menschlichen Körpers untersucht und Millionen von funktionellen genetischen Elementen gefunden. Aus diesen Studien geht hervor, dass alle Zelltypen unterschiedliche genetische Programme verwenden. Wenn man sie zusammenzählt, ist das Genom zu über 80% funktional.
In Phase 2 (2007–2012) wurden neue Technologien eingeführt wie die RNA-Sequenzierung (kurz: RNA-seq) und die Chromatin-Immunpräzipitation*-Sequenzierung (kurz: ChIP-seq). Diese Techniken ermöglichten es, das gesamte Transkriptom* von über hundert unterschiedlichen menschlichen Zellen vollständig zu kartieren* (Abb. 4). Zusätzliche Methoden ermöglichten es, die Bindungsregionen der Transkriptionsfaktoren und ihre Interaktion mit regulatorischen Proteinen im gesamten Genom zu bestimmen. Weitere Analysen zellulärer Kompartimente (Zellkern, Zytosol und subnukleare Kompartimente) ermöglichten es, die genauen Stellen der Transkripte zu bestimmen.
Im Jahr 2012 wurde mitgeteilt, dass für mehr als 80% des Genoms „biochemische Funktionen zugewiesen werden konnten, insbesondere außerhalb der gut untersuchten proteincodierenden Regionen. Fast jede DNA-Sequenz im Genom führt also irgendeine Funktion aus. Viele der entdeckten Kandidaten für regulatorische Elemente sind physisch miteinander und mit exprimierten Genen assoziiert, was neue Einblicke in die Mechanismen der Genregulation ermöglicht“ (ENCODE Consortium 2012). DNA-Regionen, die nicht für Proteine codieren, werden jetzt als nicht übersetzte Regionen („untranslated regions“, UTR) bezeichnet, da sie zwar in RNA transkribiert, aber nicht in Proteine übersetzt werden. Im Rahmen des ENCODE-Projekts wurde festgestellt, dass die UTR, gemessen an der Anzahl der in RNA-Transkripten vorkommenden Basen, viel aktiver sind: DNA-Regionen, die für Proteine codieren, werden im Durchschnitt auf fünf verschiedene überlappende und verschachtelte Weisen transkribiert*, die UTR sogar auf sieben. Zudem wurde herausgefunden, dass nicht nur ein Strang des DNA-Moleküls, sondern beide Stränge der DNA fast vollständig transkribiert werden. Damit erweisen sich früher verwendete Begriffe wie „sense“- bzw. „antisense“-Strang als irreführend.
In Phase 3 (2012–2017) machten sich die Forscher die neuesten genetischen Technologien zunutze, um Daten aus biologischen Proben zu sammeln und die regulatorischen Regionen außerhalb der Gene, in denen die meisten Variationen des Genoms von Mensch zu Mensch liegen, eingehend zu untersuchen (ENCODE consortium 2020). Anhand ihrer Daten identifizierten die Wissenschaftler etwa vier Millionen regulatorische Sequenzen („Genschalter“) in Regionen des Genoms, von denen man bisher annahm, dass sie ohne Funktion seien. Sie erweisen sich nun als entscheidend für die Kontrolle der Entwicklung und des Verhaltens von Zellen, Organen und Geweben. Darüber hinaus wurden 2.157.387 offene Chromatinregionen* gefunden und 750.392 Regionen mit modifizierten Histonen*. Diese Regionen fungieren im Sinne einer epigenetischen* Kontrolle. Bekannt als „Histoncode*“ oder „epigenetischer Code“ regulieren diese Genregionen die Zugänglichkeit des Genoms, so dass Transkriptionsfaktoren in die DNA eindringen können, um die Transkription des Gens vorzubereiten. Zudem wurden 1.224.154 Regionen beschrieben, an welche Transkriptionsfaktoren und Chromatin-assoziierte Proteine binden, 845.000 RNA-Sequenzen, die von RNA bindenden Proteinen besetzt sind, und mehr als 130.000 Interaktionen mit langer Reichweite zwischen bestimmten Genregionen (Chromatin-Loci). Die meisten dieser DNA-Abschnitte beinhalten Abschnitte, an die Proteine und RNA-Moleküle binden. Diese regulatorischen Moleküle werden dann so angeordnet, dass sie durch kooperative Interaktion die Funktion und das Expressionsniveau der proteincodierenden Gene regulieren.
Häufung im Genom von schwach-schädlichen Mutationen: Warum sind wir nicht schon längst ausgestorben?
Wissenschaftler des ENCODE-Konsortiums berichteten im Juli 2020, dass das menschliche Genom etwa vier Millionen Schalter enthält, die die Entwicklung und das Verhalten von Zellen, Organen und Geweben steuern. Sie beschreiben unter anderem 2.157.387 offene Chromatinregionen* und 750.392 Regionen mit für die DNA-Zugänglichkeit wichtigen Histon*-Modifikationen. Die bedeutendsten sind die Mono-, Di- oder Tri-Methylierung von Histon H3 an Lysin-4 (H3K4me1, H3K4me2 oder H3K4me3) und die Acetylierung von Histon-3 an Lysin-27 (H3K27ac). Diese Regionen fungieren als sogenannte epigenetische Schalter, die den Zugang zu Informationen im Genom überwachen. Sie bestimmen also, ob die DNA in RNA umgeschrieben werden kann oder nicht (Transkription). Darüber hinaus werden 1.224.154 DNA-Sequenzen und 845.000 RNA-Sequenzen von zusätzlichen regulatorischen Proteinen besetzt. Zudem wurden mehr als 130.000 DNA-Regionen gefunden, die direkte Langstreckenkontakte mit anderen Chromosomen unterhalten, damit sich die Chromosomen im Kernraum richtig positionieren können. Diese Ergebnisse bestätigen frühere Berichte von ENCODE, dass mehr als 80 Prozent des Genoms irgendeine Art von biochemischer oder interaktiver Funktion haben.
Darwinistisch gesonnene Wissenschaftler bezweifeln seit 2012 diese Erkenntnisse. Sie sind nicht damit einverstanden, dass die ENCODE-Forscher den Begriff „Funktionalität“ recht weit definieren, nämlich als jede Art von biochemischer Funktion (einschließlich Histon- und DNA-Modifikationen und DNA-DNA- und DNA-RNA-Interaktionen). Sie wissen, dass für ein Genom, das zu 80% funktional ist, die Anzahl der Mutationen (spontane, zufällige Veränderungen der DNA), die pro Generation auftreten, viel zu hoch ist. Denn Mutationen zerstören normalerweise funktionelle Elemente. Wenn mindestens 80% des Genoms funktionell sind, wären die Funktionsverluste durch Mutationen einfach zu groß. Für die Evolution werden große funktionslose Bereiche benötigt, in denen Mutationen keine negativen Folgen bewirken. Die Ergebnisse von ENCODE sprechen gegen eine nennenswerte Existenz solcher Pufferbereiche.
Die Anzahl der zufälligen Mutationen, die pro Generation auftreten, wird auf 100-1000 geschätzt (Sanford 2006). Ein Kind hat also entsprechend wesentlich mehr zufällige Mutationen in seiner DNA als seine Eltern.
Der Evolutionsbiologe H. J. Muller berechnete, dass eine zufällige Mutation pro Person bereits zu viel ist und das Genom zerfallen lässt, selbst wenn extreme Selektion solche Mutationen aus der Population entfernt (Muller 1950). Das liegt daran, dass die Menschen nicht genügend Nachkommen hinterlassen, um dem degenerativen Prozess der Mutationen entgegenzuwirken („wegzuselektieren“). Ein anderer Evolutionsbiologe, A. S. Kondrashow, ist der gleichen Meinung wie Muller. Er schrieb einen wissenschaftlichen Artikel mit dem bezeichnenden Titel: „Häufung im Genom von schwach-schädlichen Mutationen: Warum sind wir nicht schon 100 mal ausgestorben?“ (Kondrashov 1995) Auch er erkannte, dass die Anzahl der Mutationen pro Person viel zu hoch für einen aufsteigenden Evolutionsprozess ist. Er bezeichnete alle Mutationen zusammen als „Zeitbombe“; Wirbeltiere seien endgültig zum Aussterben verurteilt. Dass das Problem seitdem nicht verschwunden ist, sondern sich durch ENCODE eher verschärft hat, zeigt sein aktuelles Buch „Crumbling Genomes“ (Kondrashov 2017), was übersetzt so viel heißt wie „Zerbröselnde Genome“.
Die Information im Genom kann sich durch zufällige Mutationen dann nur noch verringern, weil auch die Auswirkungen vieler schwach schädlicher Mutationen mit der Zeit ihren negativen Einfluss geltend machen werden. Dies ist den oben erwähnten Evolutionsbiologen bekannt. Aus ihren evolutionstheoretischen Überlegungen heraus kann nur ein sehr kleiner Teil des Genoms funktional sein. Wenn nämlich fast alle Mutationen mehr oder weniger schädlich sind, könnten Genome nicht dauerhaft stabil bleiben und die Mutationen würden unwiderruflich zu einer „genetischen Kernschmelze“ führen. Evolutionstheoretiker brauchen also nicht-funktionalen „Schrott-DNA“, auf dem sich diese Mutationen ansammeln können, ohne Schaden anzurichten. Je weniger „Junk-DNA“ im Genom vorhanden ist, desto unwahrscheinlicher ist die Evolution. Eine mehrere Millionen Jahre lange Menschheitsgeschichte, wie sie gemäß Darwin’scher Theorie als Tatsache dargestellt wird, erscheint vor dem Hintergrund dieser Befunde durchaus fraglich.
Wenn es sich weiter bestätigt, dass praktisch das gesamte Genom funktional ist, wie die Ergebnisse des ENCODE-Projekts andeuten, haben praktisch alle Mutationen in der DNA eine negative Auswirkung. Es fehlen dann sozusagen die „Pufferzonen“ der Evolution. Diese Befunde widersprechen deutlich der in der Darwin’schen Lehre postulierten Höherentwicklung der Organismen und entziehen damit der Evolutionslehre im Wesentlichen ihre Erklärungskraft für die Entstehung der Artenvielfalt. Evolutionsbiologen sind daher gezwungen, die Ergebnisse von ENCODE in Frage zu stellen. Der Evolutionsbiologe Dan Graur hat deshalb vorgeschlagen, dass nicht mehr als 25 Prozent des Genoms funktional sein sollten (Graur 2017). Doch selbst bei 75% Junk-DNA kann das Genom lediglich zerfallen, nur geht es dann etwas langsamer. Sein Kommentar „Wenn ENCODE richtig liegt, dann ist die Evolution falsch“ ist bezeichnend für das Problem, das Befürworter der Darwin’schen Evolutionslehre mit einem fast vollständig funktionalen Genom haben.
Überraschungen
ENCODE bestätigte HUGOs überraschenden Befund, dass es im menschlichen Genom nur etwa 20.000 Protein-Gene gibt. Darüber hinaus fanden die ENCODE-Forscher über 37.000 RNA-Gene, die nicht in Proteine übersetzt werden, sondern regulatorische Funktionen erfüllen. Zudem sind die Gene nicht klar voneinander abgegrenzt. Sie sind nicht wie „Perlen auf einer Schnur“ (linear) angeordnet, wie man das von Bakterien kennt. Vielmehr gibt es eine verschachtelte Struktur von (mehrfach) überlappenden DNA-Segmenten, wobei typischerweise fünf Segmente (sog. Exons*) für Protein-Gene verwendet werden. Gene, die nicht für Proteine codieren (RNA-Gene), können sogar aus mehr als sieben Transkripten hergestellt werden und von verschiedenen „Genen“ stammen. Exons sind also nicht gen-spezifisch, sondern Module, die zu vielen verschiedenen RNA-Transkripten zusammengefügt werden können. Ein Exon* kann in Kombination mit anderen Exons von bis zu 33 verschiedenen Genen verwendet werden, die sich auf bis zu 14 verschiedenen Chromosomen befinden. Das bedeutet, dass ein Exon ein Strukturelement darstellen kann, das von vielen verschiedenen Proteinen gemeinsam genutzt wird. Die Kombinationsmöglichkeiten und damit die Zahl der funktionellen Produkte ist schier unendlich!
Gene werden durch Sequenzen kontrolliert, die zehn- oder sogar hunderttausende von Basenpaaren von den DNA-Sequenzen, die sie kontrollieren, entfernt sein können.
Eine zweite Überraschung war, dass Gene durch Sequenzen kontrolliert werden, die zehn- oder sogar Hunderttausende von Basenpaaren von den DNA-Sequenzen, die sie kontrollieren, entfernt sein können. Die meisten davon wurden bisher immer als Überbleibsel alter Virusintegrationen5 interpretiert, d. h. als „Junk-DNA“. Die Resultate von ENCODE belegen, dass es sich dabei um funktionelle genetische Schalter handelt. Es gibt also nicht eine einzige START-Stelle oder einen einzigen Promotor* unmittelbar vor dem ersten Exon des Gens wie bei bakteriellen Genen, sondern viele, und sie befinden sich in verschiedenen Genregionen. Im menschlichen Genom werden sie als „Enhancer“ bezeichnet, d. h. als Transkriptionsinitiatoren, die mit Promotoren vergleichbar sind, aber nicht unmittelbar vor der proteincodierenden Sequenz liegen. Die Genome von eukaryotischen Zellen unterscheiden sich also stark von denen von Bakterien. Die Schlussfolgerung, dass wir es mit zwei unterschiedlichen und separaten Designs zu tun haben, ist also nicht weit hergeholt. Im menschlichen Genom kann jedes Gen von 60-70 Schaltsequenzen angesteuert werden, welche auch auf mehrere Chromosomen verteilt sein können. Ein Gen muss daher als eine oder mehrere codierende Inseln in einem Ozean von regulatorischen Elementen verstanden werden. Das ist ein völlig neues und unerwartetes Gesamtbild unseres Genoms!
Bedeutung der Ergebnisse von ENCODE
Die Veröffentlichungen von ENCODE haben zu einem stark veränderten Blick auf das menschliche Genom geführt. Es wurde belegt, dass fast das ganze Genom in RNA übersetzt wird, und mindestens 80% davon funktional sind – im Gegensatz zur lange geglaubten Annahme, das menschliche Erbgut sei voller „Junk-DNA“. Das Genom ist ein Datenspeicher und -prozessor und einem Computer sehr ähnlich: Der Code für Protein- und RNA-Gene entspricht dem read-only-memory (ROM) des Computers, d. h. Anweisungen, um die Startbefehle für einen Computer zu speichern. Der epigenetische Code (Gen-Regulation) ist wie das random-access-memory (RAM), ein kurzfristiger Datenspeicher für Informationen, die der Computer aktiv nutzt, so dass schnell auf sie zugegriffen werden kann.

Abb. 5: Die Ergebnisse von ENCODE, einer Gruppe von Genomforschern unter der Leitung von Ewan Birney, werden von Dan Graur, einem Evolutionsbiologen und bekennenden Atheisten, heftig kritisiert. (https://www.theguardian.com/science/2013/feb/24/scientists-attacked-over-junk-dna-claim. Fotos: D. Graur: University of Houston, E. Birney: CC BY-SA 4.0)
Eine wichtige Frage ist nun: Kann das Genom ein Produkt der natürlichen Selektion bzw. Darwin‘schen Evolution sein? Denn Computer sind offensichtlich konzipierte und konstruierte Systeme und es liegt nahe, dass Zellen aufgrund ähnlicher Konstruktionsmerkmale auch geschaffen sind.
Das Autorenkollektiv von ENCODE berichtete, dass 95% der funktionellen Transkripte* (d. h. translatierte* und UTR-Transkripte mit mindestens einer bekannten Funktion) keine Anzeichen von Selektionsdruck zeigen, denn sie sind nicht merklich konserviert* (also nicht stabil) und mutieren mit einer durchschnittlichen Häufigkeit (ENCODE project consortium 2012). Das ist ein Anzeichen für neutrale Evolution: Ansammlung ungerichteter Mutationen ohne Selektion. Dies ist wirklich merkwürdig. Wenn der Mensch sich durch Mutation und natürliche Selektion aus affenähnlichen Vorfahren entwickelt haben soll, wie kann es dann sein, dass fast alle funktionellen Informationen im menschlichen Genom keinerlei Anzeichen von natürlicher Selektion aufweisen? Dies kann nur bedeuten, dass die natürliche Auslese keinen wesentlichen Beitrag zu unserer Entwicklung geleistet hat. Die Darwin’sche Evolution spielte demnach keine bedeutende Rolle bei der Entstehung des Menschen.
Die von ENCODE gemachten Erkenntnisse werden von Befürwortern des evolutionären Paradigmas angezweifelt, da sie die Gültigkeit ihres Paradigmas gründlich in Frage stellen.
Darwinisten argumentieren dagegen, dass das Nichtvorliegen einer natürlichen Selektion beweist, dass diese Sequenzen funktionslos sind und damit die Junk-DNA-Hypothese bestätigen. Tatsächlich werden alle neuen von ENCODE gemachten Erkenntnisse von Befürwortern des evolutionären Paradigmas angezweifelt, da sie die Gültigkeit ihres Paradigmas gründlich in Frage stellen (Abb. 5). In einem Vortrag an der Universität von Houston argumentierte der Evolutionsbiologe Dan Graur: „Wenn das menschliche Genom tatsächlich fast frei von ‚Junk-DNA‘ ist, wie das ENCODE-Projekt andeutet, dann kann ein langer, ungerichteter Evolutionsprozess das menschliche Genom nicht erklären.“ Und weiter sagte Graur im Sinne einer Schlussfolgerung: „Wenn ENCODE richtig liegt, dann ist die Evolution falsch.“ Nun, nach Abschluss der 3. Phase des ENCODE-Projekts, verstärkt sich die Erkenntnis: Gemäß den Befunden bezüglich des menschlichen Genoms ist die Darwin’sche Evolutionslehre falsch.
1 Manchmal besteht das Genom aus RNA (nur bei RNA-Viren).
2 Heutzutage wäre es besser, die Definition des Genoms zu ändern, da die Sequenzierung der ersten diploiden Genome ergab, dass sich die beiden elterlichen Genome um 1-3 % im genetischen Inhalt unterscheiden können. Zwischen menschlichen Populationen wurden sogar bis zu 12 % Unterschied beobachtet.
3 Nach der neuesten Version des menschlichen GENCODE (https://www.gencodegenes.org/) beträgt die Anzahl der Gene, die für Proteine codieren, 19.901.
4 Die Ansicht, dass LINEs (wie endogene Retroviren) ihren Ursprung in Viren haben, ist unhaltbar, weil es keine RNA-Viren gibt, die LINEs ähneln. LINEs haben einen einzigartigen genetischen Aufbau, und der einzige Grund, sie als Überbleibsel von RNA-Viren anzusehen, ist, dass sie ein Reverse-Transkriptase-Gen besitzen.
5 Wahrscheinlich ist es umgekehrt: RNA-Viren könnten ihren Ursprung in diesen genetischen Schaltern haben.
Studies of the chemical nature of the substance inducing transformation of pneumococcal types. Induction of transformation by a deoxyribonucleic acid fraction isolated from pneumococcus type III. J. Exp. Med. 79, 137–158.
Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816.
Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3, 318–356.
An integrated encyclopedia of DNA elements in the human genome. Nature 489, 557–574.
Perspectives on ENCODE. Nature 583, 693–698.
An Upper Limit on the Functional Fraction of the Human Genome. Genome Biol. Evol. 9, 1880–1885.
Human genome at ten: Life is complicated. Nature 464, 664–667.
Contamination of the genome by very slightly deleterious mutations: why have we not died 100 times over? J. Theor. Biol. 175, 583–594.
Crumbling Genomes. John Wiley & Sons Inc.
Our load of Mutations. Am. J. Human Gen. 2, 111–176.
www.nature.com/scitable/definition/human-genome-project-hgp-112 (abgerufen Februar 2021)
Genetic Entropy. The mystery of the genome. Feed My Sheep Foundation.
What is Life? Mind and Matter. Cambridge University Press.
Verbreitung und Ursache der Parthenogenesis im Pflanzen- und Tierreiche. Jena: Verlag Fischer, S. 165.
Themen | Kurzbeiträge | Streiflichter
Studiengemeinschaft WORT und WISSEN e.V.
Letzte Änderung: 7/2/21
Webmaster