Themen • Kurzbeiträge • Streiflichter
De novo – Gene aus dem Nichts?
Interpretationsfehler oder komplexes Genom?
von Peter Borger
Studium Integrale Journal
27. Jahrgang / Heft 2 - Oktober 2020
Seite 88 - 96
Zusammenfassung: Über 150 Jahre lang betrachteten Biologen die Evolution als einen eher konservativen Prozess: Gene sollten durch Genverdopplung und Abwandlung der Duplikate entstehen. Neue Daten zwingen die Wissenschaftler nun zum Umdenken: Ein Großteil der Gene scheint einfach so zu entstehen, ohne evolutionäre Vorschichte, gleichsam aus dem Nichts – de novo. Eine unerwartete Quelle von genetischer Information liegt in den scheinbar unnützen Regionen, die lange Zeit als „Müllhalde“ gedeutet wurden. Vieles spricht dafür, dass potenziell funktionelle Sequenzen bereits im Genom vorhanden sind und nur darauf warten, aktiviert zu werden.
• • • • • • • • •
Mit einem Stern* versehene Begriffe werden im Glossar erklärt.
Was ist ein Gen?
Ein Gen ist die grundlegende physische und funktionelle Einheit der Vererbung. Die Gene aller Lebewesen bestehen aus DNA und enthalten die notwendige Information, dass molekulare Strukturen erzeugt und diese so vernetzt werden können, dass sie funktionelle Leistungen erbringen können. In den letzten Jahrzehnten hat man erkannt, dass es bezüglich ihrer Funktion zwei grundsätzlich verschiedene Gene gibt (Abb. 1).
1. RNA-Gene: Diese Gene codieren für regulatorische RNA-Moleküle wie z. B. long non-coding RNA* (kurz: lncRNA) oder microRNA* (kurz: miRNA).
2. Protein-Gene: Diese Gene codieren für Proteine. Beide Arten von Genen haben gemeinsam, dass sie, um exprimiert* (ausgeprägt) zu werden, in RNA umgeschrieben werden müssen (Transkription*). Danach können die Produkte eines RNA-Gens weiter verarbeitet werden, aber es wird nicht in Protein übersetzt. Die Funktion liegt im RNA-Molekül selbst. Dagegen wird das RNA-Molekül (die Messenger-RNA; kurz mRNA) eines Protein-Gens in eine Aminosäuresequenz translatiert (übersetzt), die sich dann zu einem Protein faltet. Sowohl Transkription als auch Translation* werden durch zusätzliche spezifische DNA-Sequenzen gesteuert, die in das Gen eingebettet sind. Ein Gen ist somit die Summe aller DNA-Sequenzen, die zur Expression* und Regulation der codierten funktionellen Information benötigt werden. Beim Menschen variieren die Gene in ihrer Größe von einigen hundert bis zu mehr als 2 Millionen DNA-Basen. Im Juli 2020 berichtete das ENCODE-Konsortium, eine internationale Gruppe von Genom-Forschern, dass die Zahl der im menschlichen Genom vorhandenen proteincodierenden Gene etwas mehr als 20 000 und die Zahl der RNA-Gene etwa 37 000 beträgt (ENCODE Consortium 2020).Von letzteren sind schätzungsweise 16 000 lncRNA-Gene.
Kompakt
Die Entstehung neuartiger genetischer Information (neuer Gene) soll nach gängiger Erklärung das Ergebnis eines langen Prozesses sein, der mit einer Genduplikation startet. Anschließend soll eines der Duplikate (oder beide) durch Mutation und Selektion verändert worden sein. Während dieser Mechanismus immer noch als die vorherrschende Triebfeder der Evolution des Erbguts angesehen wird, zeigen neue Daten in eine andere Richtung: Eine große Anzahl von Genen scheint ohne evolutionäre Vorgeschichte, sozusagen aus dem Nichts – de novo – entstanden zu sein. Eine unerwartete Quelle genetischer Information liegt in den scheinbar nutzlosen Regionen, die lange Zeit als „Müllhalden“ („junk yards“) der Evolution interpretiert wurden. Zufällige DNA-Sequenzen zwischen funktionalen Genen sollen sich durch geringfügige Änderungen in einer Weise ändern, dass sich die codierten Aminosäureabfolgen zu Proteinen falten können. Die Wahrscheinlichkeit, dass in einer Population von zufällig generierten Sequenzen funktionale Information vorhanden ist, ist jedoch extrem gering. Ausgehend also von nichtfunktionalen (bzw. potenziell funktionalen) Sequenzen, die über Millionen von Jahren nicht exprimiert wurden, sollen dennoch funktionale Gene entstehen. Ein solches Szenario ist jedoch eher unrealistisch und wäre aus evolutionärer Sicht rätselhaft. Denn potenziell funktionale DNA-Abschnitte müssen mit regulatorischen Sequenzen verknüpft werden, damit sie exprimiert und damit genutzt werden können. Dieser Schritt ist sehr groß und daher eine Hürde, die nicht durch zufällige genetische Veränderungen genommen werden kann; er erfordert Voraussicht und Planung. Vieles deutet darauf hin, dass potenziell funktionelle Sequenzen bereits im Genom vorhanden sind und nur darauf warten, aktiviert zu werden. Die neuen Beobachtungen an de-novo-Genen implizieren, dass die Genome höherer Organismen für schnelle Veränderungen und Anpassungen ausgelegt zu sein scheinen.
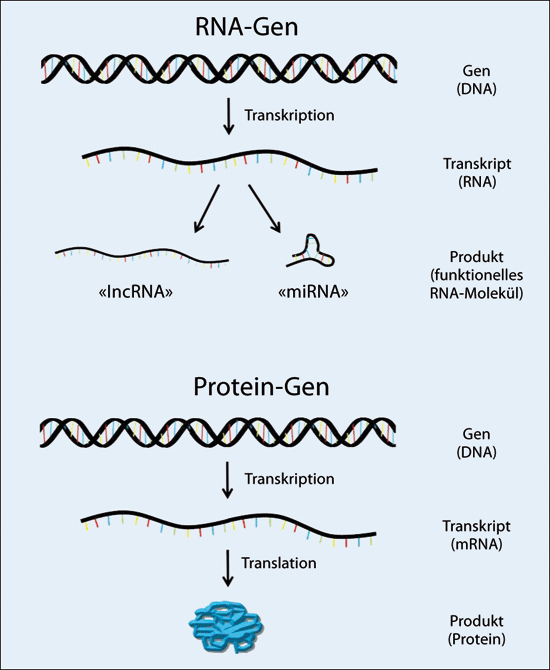
Abb. 1: In den Genomen aller Eukaryoten existieren zwei grundsätzlich verschiedene Gene, RNA-Gene und Protein-Gene. Oben: RNA-Gene werden in ein primäres RNA-Transkript umgeschrieben (Translation), das dann zu funktionsfähigen RNA-Molekülen verarbeitet werden kann. Gegenwärtig sind mehr als ein Dutzend verschiedene Arten funktioneller RNA-Moleküle beschrieben worden, darunter long non-coding RNA (lncRNA; lange Stücke nicht-codierender RNA) und Mikro-RNA (miRNA). Unten: Protein-Gene werden in ein RNA-Transkript (mRNA) umgeschrieben, das dann weiter bearbeitet und in ein funktionelles Protein übersetzt wird (Translation). Genexpression: Codes für Transkription und Translation
Um die Information eines Gens in RNA oder Protein zu exprimieren*, sind regulatorische DNA-Sequenzen erforderlich, die ähnlich wie ein Schalter funktionieren. Sie befinden sich in Bereichen, die man heute als nicht-codierende DNA bezeichnet. Verschiedene Arten von regulatorischen Elementen werden dabei unterschieden, wie z. B. Promoter*, Enhancer*, und Silencer* (Abb. 2).
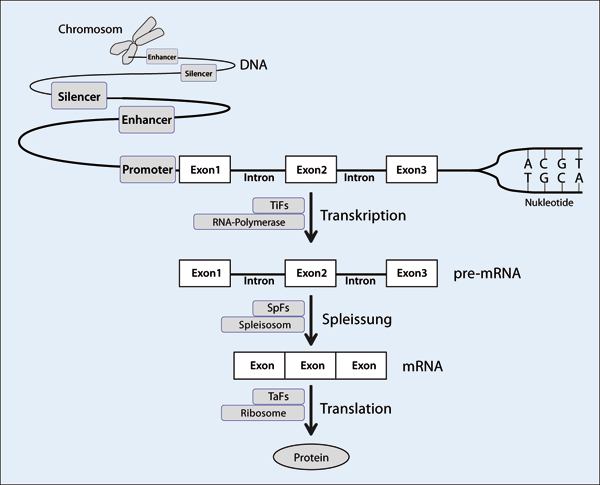
Abb. 2: Die biologische Information, die in der Reihenfolge der Nukleotide (A, T, C, G) der DNA codiert ist, befindet sich auf den Chromosomen, die die Gene enthalten. In höheren Organismen sind Gene aus Exons und Introns zusammengesetzt. Exons enthalten den Funktionscode, während Introns bei der Transkription (Umschreibung) herausgeschnitten werden. Die Transkription des Gens wird vom Promoter, oft in Zusammenhang mit sog. Enhancern und Silencern, initiiert und gesteuert. Enhancer und Silencer haben verstärkende bzw. abschwächende Wirkung auf die Transkription. Durch Kombination verschiedener Exons können ausgehend vom selben Gen unterschiedliche Proteine gebildet werden. Die Transformation der Information in der DNA zur Herstellung von Proteinen erfolgt in mehreren Schritten. Zunächst bereiten Transkriptionsfaktoren (TiFs), die an Promoter- und/oder Enhancer-Sequenzen binden, die Abstimmung auf die RNA-Polymerase vor. Die RNA-Polymerase ist das Enzym, das das Gen in ein Prä-Messenger-RNA (prä-mRNA)-Molekül transkribiert. Anschließend werden die Introns durch Enzyme, die als Spleißfaktoren (SpFs) bezeichnet werden, herausgeschnitten, was zur Bildung der reifen Boten-RNA (mRNA) führt, die nur die Transkripte der Exons enthält. Im letzten Schritt wird die mRNA in ein Protein übersetzt, was von den Ribosomen und Translationsfaktoren (TaFs) durchgeführt wird.
Der Promoter befindet sich typischerweise direkt vor dem codierenden Teil des Gens. Promoter bestehen aus einer Abfolge von Andockstellen für RNA-Polymerase* und Transkriptionsfaktoren*. Die RNA-Polymerase ist das Protein, mit dessen Hilfe die DNA in RNA umgeschrieben wird. Um die genaue Stelle zu finden, an der dieser Prozess der Transkription beginnt, müssen mehrere Transkriptionsfaktoren, welche die Bindung der RNA-Polymerase an die DNA unterstützen, zuerst an die DNA binden. Zusammen bilden sie eine Art Startplattform für die RNA-Polymerase. Die Andockstellen für die Transkriptionsfaktoren selber bestehen aus kurzen DNA-Abschnitten von 5–20 Nukleotiden oder mehr, wobei die genaue DNA-Sequenz ihre Spezifität bestimmt (d. h. welche Transkriptionsfaktoren andocken können). Es wurde festgestellt, dass im Durchschnitt 7,4 unterschiedliche Andockstellen in einem bestimmten Promoter von menschlichen Genen vorhanden sind (O‘Micks 2016).
Die am besten charakterisierten Promoter-Sequenzen, die unabhängig oder synergistisch funktionieren können, sind das TATA-Element (25 bp (Basenpaare) der Transkriptionsstartstelle mit der Sequenz TATAa/tAa/t vorgeschaltet) und ein T-und C-reiches* Initiatorelement an der Startstelle der RNA-Polymerase (Abb. 3). Dieser Promoter stellt das Haupt-DNA-Ziel für die Polymerase II dar, und die genaue Initiierung der Transkription hängt von der Komplexbildung der Polymerase II mit fünf zusätzlichen Transkriptionsfaktoren* (IID, IIB, IIF, IIE und IIH) ab (Nikolov 1997).
Enhancer kann man als Fern-Promoter betrachten. Sie befinden sich auf dem DNA-Strang vor oder nach dem codierenden Teil des Gens, das sie kontrollieren, meistens weit entfernt davon (nicht selten mehrere 100 000 bp vom Gen entfernt). Durch die räumliche Anordnung der DNA im Chromatin* kommen die Enhancer–Sequenzen der codierenden Sequenz sehr nahe. Sie bieten Bindungsstellen für Proteine, die bereits erwähnten Transkriptionsfaktoren, die bei der Aktivierung der Transkription helfen.
Silencer bieten Bindungsstellen für Proteine, die die Transkription unterdrücken. Wie Enhancer können Silencer vor oder nach dem Gen, das sie kontrollieren, gefunden werden und können sich in einiger Entfernung des Gens auf dem DNA-Strang befinden.
Den Prozess, bei dem ein mRNA-Molekül in ein Protein übersetzt wird, bezeichnet man als Translation. Es erfordert einen speziellen Code im korrekten genetischen Kontext, der auch als Kozak-Sequenz bekannt ist (Abb. 4). Bei Wirbeltieren ist die Kozak-Sequenz die Basenabfolge GCCRCCAUGG, wobei das AUG das Startcodon ist (Kozak 1999). Das R bedeutet, dass an dieser Stelle immer eine Purin-Base* (Adenin oder Guanin) sitzt. Die Kozak-Sequenz, die in allen Stämmen des Lebens einzigartig ist, wird von den Ribosomen erkannt und diese binden daran, so dass die mRNA in Protein übersetzt werden kann. Bei Prokaryoten (Bakterien und Archaebakterien) übernimmt die nicht-verwandte Shine-Dalgarno-Sequenz die Funktion der Kozak-Sequenz (Kozak 1999).
Ein Gen gleicht einem kleinen Set codierender Inseln in einem Ozean von regulatorischen Modulen.
Diese Erkenntnisse haben weitreichende Konsequenzen für das Genkonzept im 21. Jahrhundert. Bildlich gesprochen gleicht ein Gen einem kleinen Set codierender Inseln (sog. Exonen) in einem Ozean von Regulationscodes (oder: regulatorischen Modulen). Der regulatorische Teil eines Gens geht weit über den codierenden Teil hinaus, und regulatorische Module können Hunderttausende von Nukleotiden von der codierenden Region entfernt sein (Faulkner 2009). Hinzu kommt, dass ein großer Teil der Regulierungssequenzen in transponierbaren und transponierten Elementen zu finden ist, also in Bereichen, die innerhalb des Genoms verschoben werden können. Diese genetischen Elemente, die früher zur sogenannten „Junk-DNA“ gerechnet wurden, machen den größten Teil der Enhancer aus und liefern alle funktionellen Sequenzen, die zur Initiierung der Genexpression benötigt werden (Faulkner 2009; Nikitin 2019).
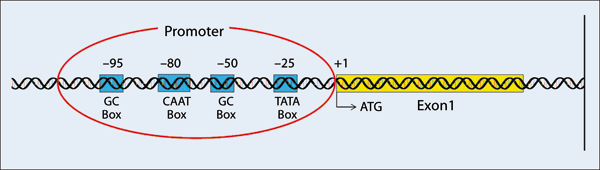
Abb. 3: Ein typischer eukaryotischer Promoter besteht aus einer TATA-Box, einer oder mehreren GC-Boxen und einer CAAT-Box. Die TATA-Box befindet sich 25–30 Basenpaare vor dem Startcodon (ATG) im ersten codierenden Teil des Gens (Exon1). Sie wird durch den Transkriptionsfaktor TFIID gebunden und bestimmt die genaue Startstelle für den RNA-Polymerase-Proteinkomplex (vgl. Abb. 2). Die GC- und die CAAT-Box werden durch den Transkriptionsfaktor* SP1 (Spezifitätsfaktor 1) bzw. CAAT-Box Transkriptionsfaktor (CBTF) gebunden, die der RNA-Polymerase helfen, die Transkription zu initiieren. Im Durchschnitt enthält ein menschlicher Promoter 7 solcher Transkriptionsfaktor-Bindungssequenzen („Box“).
Woher stammen neue Gene?
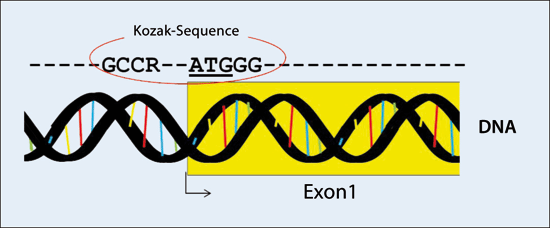
Abb. 4: Kozak-Sequenz, wie sie bei Wirbeltieren in der DNA gefunden wird. Sie findet sich unmittelbar um das Startcodon herum. Das R bedeutet, dass an dieser Stelle immer eine Purin-Base* (Adenin oder Guanin) sitzt. Die Kozak-Sequenz, die in allen Stämmen des Lebens einzigartig ist, wird von den Ribosomen erkannt und diese binden daran, so dass die mRNA in Protein übersetzt werden kann.
Die Entstehung neuer Gene wird als wichtigster Motor evolutionärer Innovationen angesehen. Die allgemeine Vorstellung ist, dass viele neue Gene durch Genverdopplung entstanden sind und dann eine Diversifizierung erfahren haben, um neue Funktionen zu erhalten. Man ging also stets davon aus, dass neue Gene entstehen, wenn vorhandene Gene sich zufällig verdoppeln, mit anderen vermischen oder mutieren. So hatte Susumo Ohno (1999) in einer richtungsweisenden Arbeit der siebziger Jahre, Evolution by gene duplication, die Auffassung vertreten: „Im strengen Sinne wird in der Evolution nichts de novo geschaffen.“
Eine große Überraschung der modernen Genomforschung ist jedoch die Beobachtung der sog. taxonomisch eingeschränkten Gene (engl: taxonomically restricted genes). Auch bekannt als Orphan-Gene* unterscheiden sie sich von anderen Genen darin, dass sie einzigartig in einem sehr engen Taxon*, in der Regel nur in einer Familie, angetroffen werden. Wissenschaftler entdeckten beispielsweise mehr als 28.000 Gene, die für Ameisen einzigartig sind und bei anderen Insekten nicht vorkommen (Simola 2013). Ebenso werden die Tintenfisch-Genome* durch Hunderte von Tintenfisch-spezifischen Genen charakterisiert (Albertin 2015). Die Genome der Singvögel besitzen einzigartige genetische Information, die für den Bau der Syrinx, dem Stimmkopf, genutzt werden.
Auch das Genom des Menschen enthält einzigartige Gene. So wurden in einer 2009 publizierten Studie 15 Gene gefunden, die nur bei Primaten, einschließlich des Menschen, vorkommen (Toll-Riera 2009). Zwei Jahre später wurden 60 einzigartige Gene für den Menschen gemeldet (Wu 2011). Im Jahr 2015 war die Zahl der bekannten menschenspezifischen Gene auf 634 angestiegen (Ruiz-Orera 2015). In allen bis dato sequenzierten Genomen machen Orphan-Gene etwa 10–30 % der identifizierten Gene aus. Neue Gene und damit neue genetische Information scheinen also plötzlich aufzutreten. Dieses plötzliche Auftreten und die funktionale Komplexität und die Integration in das Genom sprechen deutlich gegen eine schrittweise Entstehung. Diese Erkenntnis erfordert ein Umdenken: es scheint die Möglichkeit einer de-novo-Genevolution zu geben.
„Die Evolution kann durchaus etwas aus dem Nichts schaffen“ (Adam Levy).
Erst zu Beginn unseres Jahrhunderts waren Wissenschaftler in der Lage, ganze Genome eng verwandter Organismen zu vergleichen. Dies führte zu der Erkenntnis, dass Gene nicht nur plötzlich verschwinden können, sondern wie erwähnt überraschenderweise auch neu erscheinen. Während das Verschwinden von Genen vergleichsweise leicht als Verlust erklärt werden kann, erfordert das plötzliche Auftauchen neuer funktioneller Gene, bei denen eine schrittweise Entstehung nicht nachvollziehbar ist, eine plausible Erklärung.1 De-novo-Gene veranlassen Evolutionsbiologen, die konventionelle evolutionäre Sichtweise einer Genduplikation zu überdenken. De-novo-Gene, die scheinbar aus dem Nichts entstanden sind, passen nicht in das etablierte Konzept. Heute sind Evolutionstheoretiker der Meinung, dass die De-novo-Genevolution ein recht verbreiteter Mechanismus sei: Einige Studien legen nahe, dass mindestens ein Zehntel der Gene auf diese Weise entstanden ist. Manche schätzen sogar, dass mehr Gene de novo entstanden sind als durch Genduplikation. „Die Evolution kann durchaus etwas aus dem Nichts schaffen“, schreibt Levy (2019).
Glossar
Alpha-helix (-helices): Häufige Ausprägung der → Sekundärstruktur eines Proteins, ähnlich einer Wendeltreppe. Aminosäure: Baustein eines Proteins. Annotierte Gene: In der Genetik und der Bioinformatik: Funktionelle Zuordnung der Gene, die sowohl aus experimentellen Befunden als auch aus einer bioinformatischen Voraussage stammen kann. Die Annotation einer DNA-Sequenz beschreibt die genaue Lage von Exons und Introns sowie die repetitiven und funktionellen DNA-Elemente in diesen Sequenzen. Chromatin: das Material, aus dem Chromosomen bestehen. Es ist ein Komplex aus DNA und speziellen Proteinen, meist → Histonen. Coiled-coil: Strukturmotiv in Proteinen, bei dem 2 bis 7 → Alpha-Helices wie die Stränge eines Seils zusammengerollt sind. CpG-Insel: DNA-Sequenzen von etwa 1.000 Nukleotiden, besonders reich an Cytosin und Guanin. Als epigenetische Schalter steuern sie Genexpressionsprogramme. (Gen-)Expression: Ablesung, Nutzung eines Gens. Genom: das gesamte Erbgut einer Art. Histone: DNA-bindende Proteine. Long non-coding-RNA: RNA-Moleküle, die nicht wie die mRNA in eine Proteinsequenz umgebaut werden und eine Länge von über 200 → Nukleotiden aufweisen. Sie haben bestimmte biologische Funktionen, meistens als Regulatormoleküle der → Genexpression. messenger-RNA (mRNA): RNA, die bei der → Transkription gebildet wird. MikroRNA: RNA-Moleküle, die nicht wie die → mRNA in eine Proteinsequenz umgebaut werden und eine Länge von etwa 20 Nukleotiden aufweisen. Sie funktionieren als Regulatormoleküle der → Genexpression. Orphan-Gen: Gen ohne nachweisbare Homologe in anderen Abstammungslinien. Peptid: kleines Protein. Promoter: Nukleotid-Sequenz, die die regulierte → Genexpression ermöglicht. Polypeptid: Kette von Aminosäuren ohne biologische Funktion. Protein: Kette von Aminosäuren mit biologischer Funktion. Purin-Base: Ein → Nukleotid besteht unter anderem aus einer chemischen Verbindung, die als Base bezeichnet wird. Diese Base definiert die Eigenschaften der vier DNA-Buchstaben. Zwei davon sind Purinbasen (Adenin und Guanin), die beiden anderen Pyrimidinbasen (Cytosin und Thymin). Nukleotide: Bausteine der DNA (Adenin, Cytosin, Guanin, Thymin). Sekundärstruktur: Regelmäßige lokale Strukturelemente von Makromolekülen. Taxon: Einheit in der Systematik der Biologie, der entsprechend bestimmter Kriterien eine Gruppe von Lebewesen zugeordnet wird. Transkription, transkribieren: Übersetzung (Umschreibung) von DNA in (messenger-) RNA. Transkriptionsfaktor: (Komplex von) Proteinen (und manchmal auch RNA), der das Abschreiben (Transkription) eines Gens initiiert, indem er an DNA bindet und einen Ansatzpunkt für die RNA-Polymerase bildet. Transkriptionsfaktor-Bindungsstelle: Sequenz in einem DNA-Abschnitt (oder in einem Gen) für die Andockung eines → Transkriptionsfaktors, damit die → Transkription vorbereitet oder initiiert wird. Translation: Übersetzung von → messenger-RNA in ein Protein.
De-novo-Gene
Natürlich geht man nicht davon aus, dass Gene wirklich aus dem Nichts entstehen. Die Idee ist vielmehr: Orphan-Gene könnten aus Nukleotidsequenzen der DNA entstanden sein, die zuvor funktionslos waren (sog. „nicht-codierende DNA-Sequenzen“). Das ist mit de-novo-Genen gemeint. Theoretisch ist es einfach, ein RNA-Gen (wie z. B. ein lncRNA, s. o.) aus nicht-codierenden DNA-Sequenzen herzustellen, da aus jeder transkribierten DNA-Sequenz ein RNA-Molekül produziert werden kann. Dennoch benötigen die meisten lncRNAs den gleichen Transkriptionscode (s. o.) wie proteincodierende Gene und viele bestehen wie diese ebenfalls aus Exon-Inseln in einem Gen-Umfeld von Regulationssequenzen.
Es ist ebenso zu beachten, dass jede beliebige DNA-Sequenz in eine Abfolge von Aminosäuren (ein Polypeptid*) übersetzt werden kann. Dies liegt daran, dass jede Dreierfolge von DNA-Basen (Triplett aus drei Nukleotiden*) für die Proteinsynthese von Bedeutung ist. Die meisten der 64 Tripletts codieren für eine Aminosäure, während drei Tripletts bestimmen, wo die Proteinsynthese endet. Eine DNA-Sequenz, die potenziell in ein Protein übersetzt werden kann, hat Start- und Stopp-Tripletts und wird als Open Reading Frame (ORF) bezeichnet. In allen untersuchten Genomen gibt es solche ORFs, die, wenn sie die entsprechenden o. g. Transkriptions- und Übersetzungscodes enthalten, ein Polypeptid produzieren könnten. Die Expression eines ORF erfordert jedoch auch die Existenz von regulatorischen Sequenzen vor der Stelle, an der die Transkription* startet. Diese Sequenzen werden zur Steuerung des An- und Ausschaltens von Genen benötigt (s. o.). Es bräuchte zumindest eine Promotersequenz*, um die Proteine zu binden, die die mRNA-Synthese ermöglichen. Es ist aber sehr schwierig, einen funktionsfähigen Promoter von Grund auf zu evolvieren. Für das Auftreten einer einzigen Mutation an einer bestimmten Position in einem Gen zur Bildung einer neuen funktionellen Stelle wären Millionen von Jahren erforderlich (Durett 2008). Der entscheidende Punkt ist, dass irgendwelche DNA-Stücke an sich „nichts wert“ sind, sondern immer auch exprimierbar und regulierbar sein müssen. Dafür brauchen sie eine vorgeschaltete Promoter-Region, über die dieTranskription und Translation der Gene durch Transkriptionsfaktoren ein- und ausgeschaltet werden. Dennoch fanden Ruiz-Orera et al. (2015) heraus, dass die Tausenden von Transkripten, die sie als de-novo-Gene identifizierten, viele Transkriptionsfaktor-Bindungsstellen in ihren Promotern enthielten. Wie sind sie entstanden? Wie entstanden voll funktionsfähige Gene aus zuvor funktionslosen Sequenzen? Woher stammen die Regulationssequenzen in den menschlichen Genen?
Wie oben beschrieben, erfordert die Übersetzung einer DNA-Sequenz in ein RNA-Molekül einen Promoter mit Andockstellen für RNA-Polymerase und Transkriptionsfaktoren. Evolutionsbiologen vermuten, dass vereinzelte Andockstellen häufig durch Mutationen in nicht-codierenden DNA-Sequenzen entstehen können (Tautz 2011). Computersimulationen zeigen, dass zufällige Mutationen durchaus eine regulatorische Sequenz von 5 Nukleotiden erzeugen können. Mit zunehmender Länge wird die Entstehung von regulatorischen Sequenzen immer schwieriger: Eine aus 10 Nukleotiden bestehende Sequenz kommt zufällig nur noch einmal in einem Abschnitt von 1 Milliarde Nukleotiden vor. Die zufällige Konstruktion eines Promoters mit mehreren regulatorischen Sequenzen sprengt die Grenzen des Möglichen (O’Micks 2012).
Außerdem darf man von einzelnen Andockstellen nicht erwarten, dass sie direkt zur Transkription führen. Zumindest bei Eukaryonten erfordert eine aktive regulatorische Region in der Regel mehrere Bindungsstellen innerhalb eines Promoters und ein offenes, zugängliches Chromatin*, was in Kombination extrem selten auftreten dürfte. Zusammengenommen sind sowohl der ORF als auch seine regulatorischen Sequenzen für die de-novo-Entstehung eines Gens grundlegend notwendig. Im richtigen Kontext bildet die Gesamtheit dieser DNA-Elemente den so genannten Regulationscode. Genau einen solchen Code muss das de-novo-Gen erwerben, um das Gen in das Genom so zu integrieren, dass es rechtzeitig, zellspezifisch und in der erforderlichen Menge exprimiert werden kann.
Theoretisch können de-novo-Gene durch Mutationen in den Fällen entstehen, in denen die DNA umgelagert wird, ein Vorgang, der durch Deletionen (Verlust) und Einfügungen von Transposons induziert werden kann. Denn Transposons enthalten die notwendigen Expressionscodes bereits (Borger 2009). Zudem ermöglichen Transposons, dass die DNA für Transkriptionsfaktoren* zugänglich wird (Jachowicz 2017). Im evolutionären Kontext wird also der Regulationscode des Transposons für die Genexpression kooptiert (übernommen).
Viele de-novo-Gene codieren jedoch nicht für Proteine, sondern für die oben gennante lncRNA*, also für regulatorische Moleküle, die nicht in Proteine übersetzt werden und daher keine Translationscodes benötigen. Theoretisch könnten daher lncRNA-Gene in nur wenigen Schritten entstehen. Tatsächlich wird fast das ganze Genom in lncRNA abgeschrieben (The ENCODE Project Consortium 2007).
Bei codierten Proteinen ist ein zusätzliches Problem seine Funktionalität. Das Protein muss im 3-dimensionalen Raum so gefaltet werden, dass funktionelle Domänen wie z. B. Alpha-helices* und Coiled-coils* entstehen. Solche Strukturen bilden sich nur bei ganz bestimmten Mustern in der DNA-Sequenz. Die Wahrscheinlichkeit, dass in einer Population von zufällig generierten Sequenzen funktionale Information vorhanden ist, ist extrem gering. Kurze funktionelle Sequenzen von 50 Nukleotiden können in einem Pool von zufälligen Sequenzen mit einer Häufigkeit von nur einem pro 10 Milliarden gefunden werden. Funktionelle Sequenzen von 75 Nukleotiden gibt es nur noch einmal pro 1000 Milliarden (Szostak 2003). Tatsächlich besteht eine umgekehrte Beziehung zwischen Länge und Häufigkeit der Funktionalität (Graziano 2008).
Eine Forschungsarbeit der Gruppe um Diethard Tautz vom Max-Planck-Institut für Evolutionsforschung in Plön stellt dieses Ergebnis in Frage. Sie generierten zufällige DNA-Sequenzen und brachten sie in Bakterien ein. Die Forscher zeigten, dass etwa die Hälfte dieser DNA-Sequenzen das Wachstum von Bakterien verlangsamte, während etwa ein Viertel das Wachstum beschleunigte2, was darauf hindeutet, dass fast alle Zufallssequenzen funktionsfähig sind (Neme 2017). Die Daten waren jedoch nicht reproduzierbar (Knopp 2018).
Genes-in-Waiting – auf Funktion wartende Protogene?
Die ersten de-novo-Gene wurden 2006 und 2007 in Drosophila beschrieben. Sie wurden in den Hoden und den Samenflüssigkeitsdrüsen exprimiert und sind an der Fortpflanzung der Männchen beteiligt. Im Jahr 2009 berichtete die Gruppe um den Evolutionsbiologen Diethard Tautz die Neuentstehung eines lncRNA-Gens*, später Pldi genannt, das in mehreren Mäuse-Arten exprimiert wird. Bis zu 80 % der DNA-Sequenz des Pldi Gens findet man auch im Genom des Menschen, das Gen wird hier jedoch nicht exprimiert (Tautz 2009). Auf dem komplementären (gegenüberliegenden) DNA-Strang von Pldi befindet sich ein weiteres de-novo-Gen, die lncRNA Ak158810, die sich teilweise mit dem pldi-Gen überschneidet. Wie die o. g. de-novo-Gene bei Drosophila werden diese beiden de-novo-Gene in den Hoden exprimiert. Eine eingehende Analyse identifizierte verschiedene Bestandteile, darunter transponierbare Elemente (TE)*, die zur Bildung der beiden Gene beitragen – die etwa zur gleichen Zeit entstanden sein müssen (Dai 2015).
Wenn Gene wirklich auf etwas warten, was steckt dann dahinter?
Mit Hilfe der modernsten RNA-Sequenzierungstechniken entdeckten Ruiz-Orera und Mitarbeiter Tausende von jeweils einzigartigen RNA-Transkripten in Mäusen, Makaken, Schimpansen und Menschen (Ruiz-Orera 2015). Sie identifizierten 634 humanspezifische Gene, 780 schimpansenspezifische Gene und 1 300 für Hominoiden spezifische Gene, d. h. Gene, die sowohl beim Menschen als auch beim Schimpansen, nicht aber bei Mäusen und Makaken vorkommen. Insgesamt wurden somit 2 714 potenzielle de-novo-Gene gefunden. Sie waren bisher unbemerkt geblieben, weil etwa die Hälfte von ihnen kürzer als bereits annotierte* Gene sind und nur in wenigen Zelltypen und Geweben exprimiert werden. Beim Menschen codieren die meisten der 634 de-novo-Gene für lncRNA-Moleküle mit unbekannter Funktion. Von 23 dieser Gene wurde gezeigt, dass sie in Proteine übersetzt und spezifisch in Herz, Gehirn oder Hoden exprimiert werden.
Weitere Analysen zeigten, dass diese Gene durch Regulator Factor X (RFX)-Transkriptionsfaktoren* angetrieben werden, die X-Box-Promoter-Motive von 14 Nukleotiden erkennen (Konsens-Sequenz: GTNRCCNNNRG AAC)*. Motive dieser Länge sind durch zufällige Mutationen sehr schwer zu konstruieren. In einer zufälligen DNA-Sequenz, die so lang ist wie das gesamte menschliche Genom (3 Milliarden Nukleotide), würde man nicht mehr als ein solches Motiv erwarten (O‘Micks 2016). Außerdem belegt ihre kontrollierte und organspezifische Expression, dass die de-novo-Gene gut in das Genom des Menschen integriert sind. Letzteres kann mit dem Vorhandensein vieler TE zusammenhängen, die organspezifisch die Expression dieser Gene regulieren können (Pehrsson 2019). Die Wahrscheinlichkeit, dass 23 de-novo-Gene zugleich auch das gesamte Spektrum der zuvor beschriebenen regulatorischen Elemente und Transkriptions- und Translationscodes zufällig erworben haben, ist verschwindend klein. Dennoch werden im nicht-codierenden Teil des Genoms des Makaken ähnliche, aber nicht-funktionale DNA-Sequenzen beobachtet (Ruiz-Orera 2015). Dies ist der Grund weshalb sie als „Genes-in-Waiting“ („wartende Gene“) interpretiert werden (Levy 2019). Diese Tatsachen, die man aus statistischen Gründen nicht erwarten würde, bedürfen einer Erklärung. Wenn Gene wirklich auf etwas warten, was steckt dann hinter diesem Warten?
Speicher von neuen Genen?
Der in der Wissenschaftszeitschrift Nature beschriebene „Schrottplatz“ (junkyard) für Zufallssequenzen“ hat bei näherer Betrachtung eine potenzielle biologische Funktion: als Speicher von neuen Genen (Levy 2009). Dies muss den säkularen Wissenschaftler eigentlich sehr überraschen, denn eine potenzielle Funktion ist auf Zukunft gerichtet. Der anerkannte Evolutionsprozess kennt jedoch keine Zukunfts- und Zielorientierung (Teleologie). Im 21. Jahrhundert kristallisiert sich ein ganz neues Bild von den Genomen der Lebewesen heraus: Genome weisen Mechanismen auf, um Variation zu erzeugen und latente (verborgene, bereits angelegte) Information abzurufen. Genome sind nicht, wie bis vor Kurzem angenommen, statische Informationsspeicher, sondern sie sind hochdynamisch und in ständiger Veränderung begriffen. Sie können mit superdynamischen Rechnern verglichen werden, die mit sofortigen Anpassungen auf Veränderungen reagieren können, ja, diese sogar antizipieren. In Genomen, die so konzipiert sind, dass sie in der Lage sind, Variation zu erzeugen, können sich wahrscheinlich auch leicht „neue“ Gene bilden. Diese Gene sind also nicht wirklich neu, sondern werden von einem latenten, nicht-funktionalen Zustand in einen funktionalen Zustand überführt.
Auf diese Weise können Anpassung und biologischer Wandel – sogar Artbildung – einfach auf Merkmale des Genoms von Grundtypen zurückgehen, die es erlauben, neue Varietäten und Arten hervorzubringen. „Genes-in-waiting“ klingt allerdings wie bereits angesprochen nach Voraussicht, und ihre Programmierung passt eher zu einem Schöpfungskonzept (s. u.) als zu einem blinden Zufall. Sie sind ein klares Indiz dafür, dass in den Genomen der Lebewesen potenziell funktionelle Sequenzen bereits angelegt sind, die nur drauf warten, „geweckt“ zu werden. Wenn sich diese Sicht bewahrheitet, würde die Wirkung der Selektion lediglich auf Feinabstimmung reduziert. Oder, wie der britische Genetiker und Erfinder des Rekombinations-Quadrats, Reginald Punnett, vor etwa 100 Jahren notierte: „Wenn es sich herausstellen würde, das Variation im Voraus bestimmt wird, wenn es sich herausstellen würde, dass Variation Regulierung unterliegt, dann wird die Bedeutung der natürlichen Auslese zu Null reduziert“ (zit. nach Davison 2005).
Orphan-Gene, die keine Homologie mit anderen Genen aufweisen, kommen in allen Genomen vor und sind Kandidaten für de-novo-Gene. Die Entstehung von Orphan-Genen ermöglicht schnelle Artbildung, da sie neue Funktionen bereitstellen, die für Anpassungen bestimmter Abstammungslinien relevant werden können (Tautz 2011). Organismen müssen sich anpassen, wenn sich die Umgebung ändert oder wenn sie neue Lebensräume besiedeln. Diese Anpassungsfähigkeit ist gut dokumentiert, und Darwins Theorie zufolge übernimmt die natürliche Selektion hier eine wichtige Rolle. Dies gilt in den Augen der Evolutionstheoretiker auch für de-novo-Gene: „Wissenschaftler gingen lange Zeit davon aus, dass neue Gene entstehen, wenn die Evolution an alten Genen herumbastelt. Es stellt sich heraus, dass die natürliche Auslese viel kreativer ist“, wird in einer Zusammenfassung in einem aktuellen Nature-Artikel behauptet (Levy 2019). Die meisten de-novo-Gene zeigen jedoch wenige Hinweise auf Selektion. Nicht umsonst werden sie als „Protogene“ bezeichnet, als Gene, die darauf warten, geweckt zu werden (Carvunis 2012; s. o.).
Die Idee, dass alle Merkmale von Organismen das Ergebnis eines langwierigen und graduellen Selektionsprozesses auf der Basis einer natürlichen Variation sind, finden wir schon bei Charles Darwin. Als ein Mann des 19. Jahrhunderts kannte er den zugrunde liegenden Ursprung der Variation nicht, und er betrachtete Variation als Ergebnis von blindem Zufall (Darwin 1871). Für die Vorstellung, dass ein blinder Vorgang plus Selektion neue genetische Information hervorbringen kann, gibt es allerdings keine Belege. Natürliche Selektion bedeutet einfach nur unterschiedlichen Fortpflanzungserfolg. Dabei werden keine DNA-Sequenzen konstruiert, sondern solche Sequenzen beibehalten, die dem Organismus einen Fortpflanzungsvorteil verschaffen. Bevor Selektion stattfinden kann, muss die DNA-Sequenz schon vorhanden sein. Organismen können nicht darauf warten, dass die richtigen Mutationen zufällig auftreten; sie wären lange vorher bereits ausgestorben.
De-novo-Gen als Interpretationsfehler?
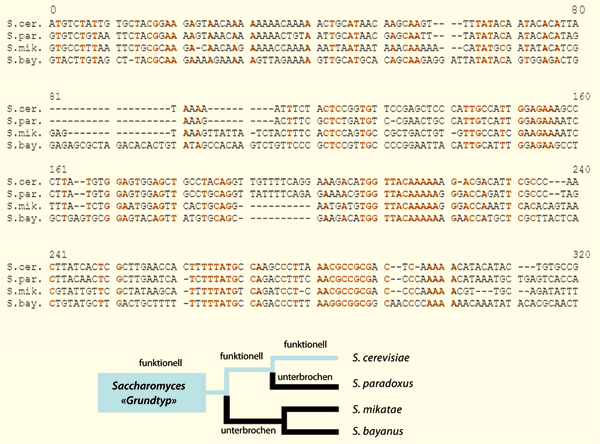
Cai et al. (2008) beschrieben eine de-novo-Evolution des Gens BSC4-Gen bei Saccharomyces cerevisiae (Bäckerhefe). Das funktionelle BSC4-Gen, das für 132 Aminosäuren codiert, kommt nur im Genom von S. cerevisiae vor, wobei Fragmente des Gens auch bei S. paradoxus, S. mikatae und S. bayanus gefunden werden. Anhand dieser Fragmente wurde ein evolutionärer Algorithmus aufgestellt, wie das BSC4-Gen schrittweise de novo entstanden sein könnte (Abb., oben). Beweist dies eine de-novo-Genevolution? Bei genauerer Betrachtung erweist sich dieses Beispiel als bloßer degenerativer Prozess, bei dem ein funktionelles Gen bei S. paradoxus, S. mikatae und S. bayanus unterbrochen und fragmentiert wurde, während es bei S. cerevisiae erhalten blieb. Das BSC4-Gen codiert für ein Protein, das an der DNA-Reparatur beteiligt ist; ein ausgeklügelter und integrierter Mechanismus, an dem Dutzende von überlappenden und daher redundanten Systemen beteiligt sind. In redundanten Systemen ist der selektive Druck auf einzelne Gene jedoch sehr schwach, wodurch sie leicht zerfallen können. Man kann sich vorstellen, dass wegen eines fehlenden Selektionsdrucks das BSC4-Gen in 3 von 4 Hefespezies3 zerfiel (Abb., unten). Die de-novo-Genevolution des BSC4-Gens ist also auf einen Interpretationsfehler zurückzuführen. Inwieweit dies auch für andere in das Genom integrierte de-novo-Gene gilt, ist unbekannt und bedarf weiterer Forschung.
Schöpfungsindiz?
Eine auf der Hand liegende Erklärung für„genes-in-waiting“ wäre, dass die Regionen des Genoms, die zwischen den (bekannten) Genen liegen, mit potenziell nützlichen DNA-Sequenzen gefüllt worden sind. Wenn dies zuträfe, wäre es ein klares Indiz für Voraussicht – und somit für Design. Denn nur denkende und handelnde Personen können zielorientiert vorgehen und für zukünftige Bedürfnisse planen. Tatsächlich codieren die meisten de-novo-Gene für Proteine, die eine alpha-helix bilden und coiled-coil-Motive besitzen (Aravind et al. 2006). Wäre das Genom ein Produkt von zufälligen evolutionären Prozessen, könnte man nicht erwarten, dass zufällig generierte DNA-Sequenzen funktionelle Proteinstrukturen erzeugen würden. Schon gar nicht wäre zu erwarten, dass das sogar häufig der Fall ist. Es ist daher sehr unwahrscheinlich, dass die nicht-codierenden DNA-Sequenzen, die zu codierenden werden können und somit neue funktionale Proteine produzieren sollen, zuvor völlig zufällige Anordnungen von Nukleotiden waren. In Anbetracht dessen, was wir derzeit über die Genexpression wissen, würden bisher bekannte, rein natürliche Mechanismen, die nur auf Zufall und Naturgesetzen beruhen, nicht funktionieren, um neue Gene zu erzeugen. Mit eingebauten potenziell funktionellen Genen scheint das Genom jedoch so angelegt zu sein, dass Variation, Veränderung und die Entstehung neuer Arten vorprogrammiert sind.
1 Die Orphan-Gene sprechen klar gegen die Idee einer gemeinsamen Abstammung. Da jedoch alle Merkmale von Organismen als eine Kombination aus gemeinsamer Abstammung plus Veränderung interpretiert werden können, ist die Evolutionstheorie gleichsam gerettet. Einerseits werden Merkmale, die zwei verschiedene Organismen gemeinsam haben, als Beweis für eine gemeinsame Abstammung interpretiert. Zum anderen werden einzigartige Merkmale, die gegen gemeinsame Abstammung sprechen, als Veränderungen interpretiert – und als solche werden sie auch als Beleg für Evolution gewertet. Mit anderen Worten, es spielt hier keine Rolle, was wir beobachten: Evolution ist immer wahr.
2 Die Wachstumsrate eines Bakteriums sagt allerdings nichts über die Funktionalität eines Proteins aus. Vielmehr wechselwirken solche Sequenzen mit genetischen Netzwerken, die für Wachstum verantwortlich sind, und stören diese.
3 Aus der Sicht der Redundanz kann das BSC4-Gen aus dem Genom der Bäckerhefe entfernt werden, ohne größere Probleme zu verursachen.
The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 524, 220–224.
Comparative genomics and structural biology of the molecular innovations of eukaryotes. Curr. Opin. Struct. Biol. 16, 409–419.
Darwin Revisted – or how to understand biology in the 21st century. Scholars’ Press.
Proto-genes and de novo gene birth. Nature 487, 370–374.
The de novo sequence origin of two long non-coding genes from an inter-genic region. BMC Genomics 14(Suppl 8), S6.
Die Abstammung des Menschen und die geschlechtliche Zuchtwahl (engl. :The Descent of Man, and Selection in Relation to Sex). John Murray, UK,
A Prescribed Evolutionary Hypothesis. Riv. Biol. 98, 155–165.
Waiting for two mutations: with applications to regulatory sequence evolution and the limits of Darwinian evolution. Genetics180, 1501–1509.
Identification and analysis of functional elements in 1 % of the human genome by the ENCODE pilot project. Nature 447, 799–816.
Perspectives on ENCODE. Nature 583, 693–698.
The regulated retrotransposon transcriptome of mammalian cells. Nature Genetics 41, 563–571.
Selecting folded proteins from a library of secondary structural elements, J. Am. Chem. Soc. 130, 176–185.
LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo, Nature Genetics 49, 1502–1510.
Functional information: Molecular messages Nature 423, 689.
No beneficial fitness effects of random peptides. Nature Ecol. Evol. 2,1046–1047.
Initiation of translation in prokaryotes and eukaryotes. Gene 234, 187–208.
Genes from the junkyard. Nature 574, 314–316.
Random sequences are an abundant source of bioactive RNAs or peptides. Nature Ecol. Evol. 1:0127.
Retroelement-linked transcription factor binding patterns point to quickly developing molecular pathways in human evolution. Cells 8, pii: E130.
RNA polymerase II transcription initiation: A structural view. Proc. Natl. Acad. Sci. 94, 15–22.
Evolution by gene duplication. Springer.
Promoter evolution is impossible by random mutations. J. Creat. 30, 60–66.
The epigenomic landscape of transposable elements across normal human development and anatomy. Nature Commun. 10:5640.
De novo ORFs in Drosophila are important to organismal fitness and evolved rapidly from previously non-coding sequences. PLoS Genet. 9(10):e1003860.
Origins of de novo genes in human and chimpanzee. PLoS Genet 11(12):e1005721.
Social insect genomes exhibit dramatic evolution in gene composition and regulation while preserving regulatory features linked to sociality. Genome Research 23,1235–1247.
The evolutionary origin of orphan genes. Nat. Rev. Genet. 12, 692–702.
The proportion of polypeptide chains which generate native folds – part 3: Reusing existing secondary structures. J. Creat. 25,102–105.
Origin of Primate orphan genes: A comparative genomics approach. Mol. Biol. Evol. 26, 603–612.
De novo origin of human protein-coding genes. PLoS Genet. 7: e1002379.
Themen | Kurzbeiträge | Streiflichter
Studiengemeinschaft WORT und WISSEN e.V.
Letzte Änderung: 6/28/21
Webmaster