Themen • Kurzbeiträge • Streiflichter
Artübergreifende wiederkehrende Mutationen
Oder: Die Illusion der Verwandtschaft
von Peter Borger
Studium Integrale Journal
26. Jahrgang / Heft 2 - April 2019
Seite 77 - 85
Zusammenfassung: Gemeinsame Sequenzunterschiede in der DNA verschiedener Arten und höherer Taxa gelten als klare Belege für die Existenz gemeinsamer Vorfahren, bei denen eine bestimmte Mutation aufgetreten ist, die an die Nachfahren vererbt wurde. Voraussetzung für diese Schlussfolgerung ist die Zufälligkeit aller Mutationen. Allerdings mehren sich Befunde, dass ein Teil der Mutationen an bevorzugten Stellen im Genom – sogenannten Hotspots – auftritt. Das hat weitreichende Konsequenzen für die Deutung von Abstammungshypothesen.
• • • • • • • •
Einleitung
Mutationen, die in DNA-Sequenzen der Keimbahn auftreten, können von einer Generation zur nächsten weitergegeben werden. Da solche Mutationen somit an die Nachkommen vererbt werden, stellen sie ein ideales Werkzeug dar, um die genetische Verwandtschaft zwischen den Arten zu untersuchen. Der aktuelle Konsens ist, dass alle Mutationen nach dem Zufallsprinzip auftreten und daher nur einmal vorkommen – mit Ausnahme einiger „Hotspots“*1. Die gängige Meinung ist: Mutationen sind weder vorhersehbar noch hängen sie zusammen mit Verhalten, Lebensstil oder Umweltumständen. Diese Sichtweise wird von Futuyma in seinem weltweit verwendeten evolutionären Lehrbuch wie folgt zusammengefasst:
„Mutationen sind in zweierlei Hinsicht zufällig. Erstens: Obwohl wir die Wahrscheinlichkeit vorhersagen können, dass eine bestimmte Mutation auftreten wird, können wir nicht vorhersagen, welche von einer großen Anzahl an Genkopien die Mutation durchlaufen wird. Zweitens … ist Mutation zufällig in dem Sinne, dass die Wahrscheinlichkeit, dass eine bestimmte Mutation auftritt, nicht davon beeinflusst wird, ob sich der Organismus in einer Umgebung befindet, in der diese Mutation vorteilhaft wäre, oder ob das nicht der Fall ist“ (Futuyma 2005).2
Mit einem Stern* versehene Begriffe werden im Glossar erklärt.
Obwohl Futuymas zweite Art der Zufälligkeit nicht mehr wissenschaftlich haltbar ist, da gezeigt wurde, dass der Organismus in der Lage ist, gezielte Mutationen als Reaktion auf Umweltveränderungen zu induzieren (Spetner 1997; Caporale 2003; Bauer 2008; Borger 2008), werde ich mich hier auf die erste Art der Zufälligkeit konzentrieren. Denn sie ist eine der Säulen des Darwin‘schen Paradigmas und eine der wichtigsten Voraussetzungen phylogenetischer Rekonstruktionen. Werden alle Mutationen als zufällige Kopierfehler in DNA-Sequenzen eingebracht, wäre jedes Alignment* von Mutationen in DNA-Sequenzen ein sehr starkes Argument für eine gemeinsame Abstammung, insbesondere dann, wenn gleiche Mutationen in inaktivierten Genen verschiedener Arten vorkommen. Genau diese Situation treffen wir beim GULO-Pseudogen* bei Primaten an, so beim Orang-Utan, Gorilla, Schimpansen und Menschen. Es ist deswegen verständlich, dass das GULO-Pseudogen immer wieder als endgültiger Beweis für eine Makro-Evolution vom Affen zum Menschen angeführt wird. Wenn alle Mutationen nur ein zufälliges Phänomen wären, würde das GULO-Pseudogen tatsächlich die Makroevolution beweisen. Aber wie willkürlich bzw. zufällig sind genetische Mutationen wirklich?
Kompakt
Vor dem Aufkommen der Molekularbiologie basierten evolutionäre Abstammungslinien überwiegend auf morphologischen und ontogenetischen Merkmalen, also auf dem Bau und der individuellen Entwicklung der Arten. Heutzutage dagegen beruhen die meisten Modellierungen auf molekulargenetischen Sequenzanalysen (Abfolgen des Erbmoleküls DNA). Überraschenderweise stellt sich heraus, dass gleichartige Mutationen oft in verschiedenen Arten, die nicht näher miteinander verwandt sind, auftreten. Dieses als Konvergenz bezeichnete Phänomen deutet auf ein unerwartetes Phänomen bei Mutationen hin.
Die neuesten Forschungsergebnisse zeigen, dass ein hoher Prozentsatz der Mutationen an sogenannten DNA-Hotspots aufzutreten scheint, d. h. an Stellen in der DNA, die eher Mutationen zulassen. Diese Tatsache hat schwerwiegende Auswirkungen auf die Evolutionsbiologie, insbesondere auf Schlussfolgerungen, die sich auf die Zufälligkeit von Mutationen stützen. Bestimmte molekulargenetische Daten wie z. B. mutmaßlich vererbte Fehler in Pseudogenen schienen bisher ein starkes Argument gegen eine getrennte Erschaffung von Grundtypen zu sein. Dieses Argument wird durch die Erkenntnis entkräftet, dass Mutationen sich nicht nur zufällig in einer DNA-Sequenz ansammeln. Die Mutationen an Hotspots sind nämlich phylogenetisch nicht informativ, d. h. sie sind keine Hinweise auf gemeinsame Vorfahren, sondern sind unabhängig entstanden und dann nur noch als Konvergenzen zu verstehen.
Beispiele für wiederkehrende Mutationen
Dass Mutationen nicht zufällig in DNA-Sequenzen auftreten, zeigt sich z. B. bei Mutationen im 1G5-Gen der Taufliegen Drosophila melanogaster und Drosophila simulans, zwei verschiedenen Arten von Taufliegen (Schmid & Tautz 1997; Abb. 1). In diesem Gen wurden 75 Mutationen vorgefunden, meistens Punkt-Mutationen*. Es wurde festgestellt, dass sie gleichmäßig über die kodierenden und nicht-kodierenden Bereiche des Gens verteilt waren. Die Autoren kamen daher zu dem Schluss, dass sich die 1G5-Gene in beiden Organismen neutral entwickeln. Das bedeutet, dass die Gene einfach Mutationen anhäufen, ohne dass die natürliche Selektion eine Rolle spielt. Es war den Autoren aber entgangen, dass die Mutationen im 1G5-Gen nicht zufällig in der Nukleotidsequenz des Gens angetroffen werden. Abb. 1 zeigt, dass es im betreffenden Gen von D. melanogaster nur eine beschränkte Anzahl an Positionen gibt, in denen Mutationen stattfinden. Liest man die Sequenzen von oben nach unten, sieht man deutlich ein Muster. Das Besondere ist, dass das Muster der Mutationen, d. h. die Verteilung, wie sie in den Sequenzen der verschiedenen Taufliegen vorkommen, die Illusion einer gemeinsamen Abstammung hervorruft. Das soll im Folgenden genauer erläutert werden.

Abb. 1: Nicht-zufällige Mutationen in der Fruchtfliege Drosophila melanogaster. Vor 1875 gab es keine Populationen von D. melanogaster in Kanada und in den anderen Regionen Nordamerikas und vor 1900 keine in Australien. Die heutigen Populationen von D. melanogaster auf dem amerikanischen und australischen Kontinent gehen auf Invasionen der jüngsten Zeit zurück. Die Fliegen stammen hauptsächlich aus eurasischen Populationen (die relativ homogen sind) und ein kleiner Teil aus afrikanischen Populationen. Eine australische Population hätte z. B. keine Nische in Russland übernehmen können, weil es damals gar keine australische Population gab (oder japanische oder mexikanische Population; es gab Drosophila auch nicht in Japan vor den 1960er-Jahren und auch nicht in Mexiko vor etwa den 1900er-Jahren). Drosophila entwickelte sich in Afrika, bevor sie in Eurasien einwanderte. Von dort aus brachten die Menschen Drosophila unabsichtlich mit ihren Schiffen und Flugzeugen nach Amerika und Australien. Die fett gedruckten Mutationen (Positionen 498, 835 und 861) können nur als nicht-zufällige „Hotspot“-Mutationen verstanden werden.
Vor 1875 gab es D. melanogaster noch nicht auf dem nordamerikanischen Kontinent. Vor 1900 kam sie auch nicht in Australien vor, während sie Japan erst etwa 1960 erreichte. Heutzutage wird sie weltweit angetroffen. Die heutigen Populationen von D. melanogaster in Nord- und Südamerika, in Australien und Japan sind daher Einwanderer in jüngerer Zeit, die von europäischen und asiatischen Populationen abstammen. Wenn wir die Sequenzen in Abb. 1 studieren, fällt auf, dass eine italienische Population sich in Amerika angesiedelt hat. Zuerst in den Vereinigten Staaten (Dmel12) und später in Peru (Dmel9). Dies können wir anhand von fünf identischen Mutationen feststellen. Diese Mutationen trifft man nur in diesen drei Populationen an. Die peruanische Population trägt außerdem eine ähnliche Mutation, wie man sie in der japanischen Population antrifft (Dmel8), nämlich das A auf Position 835. Darüber hinaus kann man schließen, dass zypriotische Taufliegen die Populationen in der ehemaligen UdSSR (Dmel3) und im Irak (Dmel6) gegründet haben, ebenso die von Kanada (Dmel4) und den USA (Dmel11). Weiter fällt auf, dass eine australische Population (Dmel2) genau dasselbe T auf Position 498 besitzt wie die Populationen von Peru (Dmel9) und den USA (Dmel12). Schließlich sehen wir noch dieselbe Mutation auf Position 861 in australischen (Dmel3) und amerikanischen Taufliegen (Dmel10).
Diese Tatsachen legen nahe, dass Mutationen nicht nur zufällig in einem DNA-Abschnitt auftreten können. Ein Teil der Mutationen, die normalerweise als Argument für gemeinsame Abstammung aufgefasst werden, könnte deswegen wiederkehrend sein, also wiederholt in gleicher Art auftreten. Wenn wir keine geographischen Daten gehabt hätten, so wären wir geneigt, einen rezenten, gemeinsamen Vorfahren für Dmel1, -4, -5, -6, -11 und -13 vorauszusetzen, weil sie alle die genau gleiche DNA-Sequenz aufweisen. Wir würden ebenfalls schließen, dass Dmel7, -9 und -12 einen rezenten Vorfahren haben. Und dasselbe trifft für Dmel3 und -4 zu. Wenn die Erzeugung von Mutationen im Erbgut von Lebewesen durchweg ein zufälliger Prozess wäre, so wäre diese Folgerung berechtigt. Die variablen Positionen im 1G5-Gen legen jedoch nahe, dass wir es hier mit gleichartigen, wiederkehrenden Mutationen zu tun haben. Zugleich stellt sich heraus, dass sich an diesen Positionen die DNA-Basen (Nukleotide) nicht beliebig verändern, sondern durch einen ähnlichen Buchstaben ersetzt werden. Eine wichtige Frage ist jetzt: Sind solche non-random mutations ein allgemein vorkommendes Phänomen oder nur eine Ausnahme? Sollten sie allgemein vorkommen, so ist es schwierig, einen Unterschied zwischen gemeinsamer Abstammung und einem gemeinsamen Mutations-Erzeugungs-Mechanismus abzuleiten.
Bei Menschen wurden bisher wiederkehrende Mutationen sowohl in der Kern-DNA (Hernandez-Pacheco 2017) als auch in mitochondrialer DNA (mtDNA) festgestellt (Ruiz-Pesini et al. 2004).3 Sogar eine gleichartige Verlustmutation (Deletion), welche die mtDNA um neun DNA-Basen verkürzt, entstand mehrmals unabhängig in Afrika und Asien. Da die Mutation in Afrika nur bei den Khoi-San, den Pygmäen und den Bantus gefunden wurde, war sie nicht auf einen gemeinsamen Vorfahren oder auf Auswanderung zurückzuführen. Darüber hinaus wurde die gleiche Mutation ebenso in Südostasien, Polynesien und in Nord- und Südamerika beobachtet (Soodyall et al. 1996). Die Genombiologie des 21. Jahrhunderts führt somit in eine neue „undarwinsche“ Welt, in der manche Mutationen vorhersehbar und sogar berechenbar sein könnten. Wenn Populationen von Stichlingen in neue Lebensräume gelangen, finden Anpassungen oft auf sehr ähnliche, ja sogar vorhersehbare Weise statt, indem sie die hinteren Beckenflossen verlieren. Auf molekularer Ebene ist dieser Verlust (auch bekannt als „pelvic reduction“) meist mit einer Enhancer-Sequenz* im Genom der Fische verbunden. Diese Sequenz liegt in einem Bereich des Genoms, der aufgrund seines hohen Thymin-Guanin-Gehalts stark zum doppelsträngigen DNA-Bruch neigt. Der Bruch, der immer an dieser Stelle auftritt, legt nahe, dass das Genom gleichsam auf Umweltveränderungen vorbereitet ist, damit durch wiederkehrende Mutationen schnelle Anpassungen ermöglicht werden (Xie et al. 2019).
Umweltinduzierte wiederkehrende Mutationen
Im Jahr 2002 erschien eine interessante Studie der Universität Münster, die aufzeigte, dass wiederkehrende Mutationen auch ein generelles Phänomen beim Menschen sein könnten. Die Studie hatte das Ziel, die Auswirkungen einer erhöhten natürlichen Radioaktivität auf die Mutationsfrequenz des DNA-Moleküls festzustellen. Natürliche Radioaktivität ist energiereiche ionisierende Strahlung und entsteht überall in der Natur, wo instabile Atome sich unter Energieabgabe (radioaktive Strahlung) in stabile Atome umwandeln. Die dabei freiwerdende Energie in Form von Beta- und Gammastrahlen nennt man Radioaktivität. Diese Strahlung kann vererbbare Mutationen in DNA-Sequenzen auslösen. Nach allgemein akzeptierter Auffassung kommen solche strahlungsbedingten Mutationen zufällig verteilt in einer DNA-Sequenz vor. Man kann daher nicht vorhersagen, an welcher Stelle der DNA-Sequenz die Mutationen auftreten werden, wenn ein Organismus der radioaktiven Strahlung ausgesetzt ist. Die Studie lieferte jedoch den ersten empirischen Beweis für den nicht-zufälligen Charakter der strahlungsinduzierten Mutationen, als die Mutationen einer menschlichen Population von Kerala, einer Region im äußersten Südwesten des indischen Kontinents, genauer analysiert wurden. Generationen von Menschen verdienten hier ihr Brot als Fischer. Sie trockneten ihre Netze an den exotischen Stränden des Indischen Ozeans, an denen feiner schwarzer Kristallsand, der sogenannte Monasit, vorkommt. Da Monasit Thorium enthält, ist Kerala die Region mit der höchsten natürlichen Radioaktivität der Welt. In Kerala gibt es auch Regionen mit einer normalen radioaktiven Hintergrundstrahlung, weswegen dieses indische Gebiet ein einmaliges natürliches Labor darstellt. Das Münsteraner Team nahm für seine Untersuchungen nahezu tausend Blutproben von Probanden, die seit Generationen entweder das stärker radioaktive Gebiet oder das normale Hinterland bewohnten. Die Proben wurden im deutschen Labor auf das Vorhandensein von Mutationen in der mitochondrialen DNA analysiert. In den Proben des stärker radioaktiven Gebiets wurden 22 Punktmutationen gefunden; in der Kontrollgruppe nur eine. Dass eine hohe natürliche Radioaktivität zu einer größeren Mutationsfrequenz führen würde, hatten die Forscher zwar erwartet; was sie aber nicht vorhergesehen hatten, war, dass diese Mutationen nahezu alle auf Hotspots angetroffen wurden:
„Unglaublicherweise fördern diese radioaktiven Umstände Mutationen auf Positionen, die seit mindestens 60.000 Jahren evolutionäre Hotspots sind“ (Foster et al. 2002).
Mit den 60.000 Jahren spielt der Autor darauf an, dass die gleichen Mutationen bereits in der mtDNA der australischen Aborigines gefunden wurden, die nach gängiger evolutionärer Auffassung seit mindestens 60.000 Jahren von den in Kerala lebenden Menschen getrennt sind. In einem Gebiet mit erhöhter natürlicher Radioaktivität finden wir also erneut Hinweise auf wiederkehrende Mutationen, obwohl dies nicht der Zweck der Studie war. Das Ergebnis war eine Überraschung. Man kann daraus schließen: Da radioaktive Strahlung nicht beliebig verteilte Mutationen hervorruft, könnte dies auch auf Mutationen zutreffen, die durch UV-Strahlung oder oxidativen Stress hervorgerufen werden. Wenn es sich so verhielte, könnte vorhergesagt werden, dass sich Mutationen bevorzugt in bestimmten Bereichen ereignen, nämlich den Hotspots. Das würde die bisherigen Vorstellungen zur molekularen Evolution gehörig infrage stellen, da solche Mutationen die Illusion einer gemeinsamen Abstammung bewirken.
Ein ähnliches Beispiel ist eine genetische Anpassung im Genom der Bewohner der Quebrada-Camarones-Region in der chilenischen Atacama-Wüste, die vor Arsenvergiftung schützt. Als vor mehreren Tausend Jahren die Wüste ihre Heimat wurde, begannen die Ur-Chilenen, Wasserquellen mit hoher Konzentration an Arsen zu nutzen. Die Arsenverunreinigung dieser Quellen übersteigt manchmal ein Milligramm pro Liter, was mehr als 100-mal höher ist als nach den Regeln der Gesundheitsorganisationen der Welt zulässig. Da es praktisch keine alternativen Wasserquellen gibt, waren die in der Wüste lebenden Ureinwohner gezwungen, das giftige Wasser zu trinken. Unerwartet gewöhnten sie sich schnell an das giftige Wasser, was sich als Folge einer Mutation im AS3MT-Gen herausstellte, wodurch der Körper Arsen abbauen kann. Das adaptierte Gen bewirkt, dass die Bewohner von Quebrada-Camarones Arsen viel schneller detoxifizieren (unschädlich machen) und so in der Wüste überleben können. Frühere Studien fanden ähnliche Mutationen im AS3MT-Gen, die zu einem verbesserten Arsen-Metabolismus in Vietnam und Argentinien beitragen (Apata et al. 2017).
Mechanismen für wiederkehrende Mutationen
2017 wurden weitere Indizien dafür präsentiert, dass Mutationen an vorhersehbaren Positionen im Genom auftreten können. Eine seltsame Beobachtung der modernen Genombiologie ist, dass manchen Organismen essenzielle (lebenswichtige) Gene zu fehlen scheinen. Eine Forschungsgruppe um Adam Hargreaves in Oxford, England, beschreibt diese Gene als „dunkle DNA“ („Dark DNA“). Die von ihnen untersuchten Gene sollten eigentlich existieren, weil sie eine besonders wichtige Funktion erfüllen und lebensnotwendig sind. Als sie nach einem Gen namens Pdx1 suchten, das die Sekretion von Insulin steuert, fanden sie heraus, dass es fehlte, ebenso wie 87 andere Gene in der Umgebung von Pdx1. Einige dieser fehlenden Gene, einschließlich Pdx1, sind essenziell und ohne sie kann ein Tier nicht überleben. Die Forscher entdeckten, dass die DNA-Sequenzen dieser Gene sehr reich an den Nukleotiden* Guanin (G) und Cytosin (C) sind, während sie weniger von den beiden anderen DNA-Buchstaben, Adenin (A) und Thymin (T), enthielten. Da GC-reiche Sequenzen für bestimmte DNA-Sequenzierungstechnologien Probleme verursachen, ist die richtige Reihenfolge viel schwieriger nachzuweisen (daher „dunkle DNA“4. Als sie die Region genauer untersuchten, stellten die Forscher fest, dass ein Teil der Gene viel mehr Mutationen aufwies als erwartet. Interessanterweise waren die Gene mit Hotspots übersät, sodass sie stark mit GC-DNA angereichert und so häufig mutiert waren, dass sie mit Standardmethoden schwer nachzuweisen sind. Auf seiner Website formulierte Hargreaves als Schlussfolgerung, dass es sich um einen Mechanismus handeln muss. Er schreibt:
„Hotspots mit hoher Mutationsrate innerhalb eines Genoms bedeuten, dass Gene an bestimmten Orten eine höhere Mutationswahrscheinlichkeit haben als andere. Dies bedeutet, dass solche Hotspots ein unterschätzter Mechanismus sein könnten, der auch die Richtung der Evolution beeinflussen könnte, was bedeutet, dass die natürliche Selektion möglicherweise nicht die einzige treibende Kraft ist“ (Hargreaves 2017).
Die wiederkehrenden Mutationen waren zuvor vermutlich nicht bemerkt worden, weil nicht genügend Datenmaterial zur Verfügung stand.
Wie könnten solche Mechanismen aussehen? Indem die Evolutionsbiologie a priori von der These ausgeht, dass adaptive genetische Änderungen nur das Ergebnis zufälliger von der Selektion begünstigter Mutationen sein können, wurden systematische Studien weitgehend vernachlässigt, die die These von wiederkehrenden Mutationen untermauern könnten. Die wenigen Daten, die uns zur Verfügung stehen, stammen von einer Studie mit E. coli-Bakterien. Zwei große DNA-Abschnitte wurden analysiert. Die Analyse des ersten Abschnitts ergab, dass 63 % der 293 unabhängig aufgetretenen Mutationen auf nur 19 verschiedene Hotspots* verteilt waren. Im zweiten Abschnitt machten die Forscher neun Hotspots aus, an denen der größte Teil der beobachteten 120 Mutationen lokalisiert war. In einem vergleichbaren Experiment fand man 70 % der Punktmutationen auf nur zwei extremen Hotspots, die restlichen 475 Mutationen waren über 91 Positionen gleichmäßig verteilt (Maki et al 2002). Die Häufung von Mutationen an bestimmten Genorten in Bakterien ist dementsprechend kein zufälliges Phänomen; vielmehr spielen bislang unbekannte Mechanismen eine Rolle, die man nur nachweisen kann, wenn man eine ausreichend große Anzahl von Individuen analysiert. Genau das macht das Exom Aggregation Consortium seit 2017. Diese internationale Gruppe von Wissenschaftlern versucht die Daten aus einer Vielzahl von umfangreichen Sequenzprojekten zusammenzutragen, damit sie für die breitere wissenschaftliche Gemeinschaft zur Verfügung stehen. Der öffentlich zugängliche Datensatz, den das Konsortium auf seiner Website bereitstellt, umfasst DNA-Sequenzen von mehr als 60.000 nicht verwandten Personen (http://exac.broadinstitute.org). Diese groß angelegte Datenbank wurde für medizinische und funktionell-biologische Zwecke erstellt und ist der bisher größte Katalog der menschlichen genetischen Variation bzw. Vielfalt. Der Katalog enthält durchschnittlich eine Variante pro acht Basen des transkribierten Genoms und liefert den bislang überzeugendsten direkten Nachweis für wiederkehrende Punktmutationen. Etwa die Hälfte aller menschlichen Tumore gehen auf Mutationen an mittlerweile vorhersehbaren Stellen zurück, da sie auf einem oder mehreren Hotspots auftreten (Chang et al. 2016).
Während diese Mutationen bisher als zufällige Ereignisse angesehen wurden, die durch DNA-Replikationsfehler oder durch DNA-schädigende Mittel induziert wurden, führte die Genomsequenzierung zur Entdeckung einer erkennbaren Mutations-Signatur bei vielen menschlichen Krebsarten. Viele dieser Mutationen beschränken sich auf bestimmte DNA-Motive, die der Aktivität einer Familie von DNA-Deaminasen, der AID/APOBEC-Enzyme, zugeschrieben werden. Diese Enzyme sind Schlüsselakteure in der natürlichen und adaptiven Immunität*, können aber zur Krebsentwicklung und zu einem klonalen Entwicklungsgang von Krebs beitragen, indem sie genomische Schäden aufgrund ihrer DNA-deaminierenden Aktivität* induzieren. Daher liefert diese Protein-Familie, zumindest teilweise, eine Erklärung und einen biologischen Mechanismus für die Beobachtung von nicht-zufälligen, wiederkehrenden Mutationen (Rebhandel et al. 2015). Es ist davon auszugehen, dass die wiederkehrenden Mutationen zuvor nicht bemerkt worden waren, weil für die bisher veröffentlichten Studien nicht genügend Datenmaterial zur Verfügung stand. Nur die Analyse großer Datenmengen zeigt, dass Mutationen oft an vorhersehbaren Stellen auftreten. Eine solche Erkenntnis hat enorme Konsequenzen für Evolutionstheorien, insbesondere wenn es um die Erklärung von artübergreifenden Mutationen geht.
Glossar
Adaptive Immunität: Die adaptive Immunität ist gekennzeichnet durch die klonale Expansion der Lymphozyten und durch das immunologische Gedächtnis. Es ist adaptiv, weil es nicht angeboren (oder natürlich) ist und sich an die Eindringlinge anpasst. Alignment: „Ausrichtung“, bezeichnet den methodischen Vergleich zweier oder mehrerer DNA-Sequenzen. Es wird in der molekularen Phylogenie verwendet, um evolutionäre Verwandtschaften zu bestimmen. Catarrhini: Unterordnung der Primaten, zu der auch der Mensch gehört. Die andere Unterordnung sind die Platyrrhini. DNA-deaminierende Aktivität: die chemische Abspaltung einer Aminogruppe von der DNA. Enhancer-Sequenz: DNA-Abschnitt (DNA-Code), der die Transkription („Umschreibung“ in mRNA, genetischer Output) eines Gens verstärkt. Exon: Abschnitt der DNA, welcher in eine Aminosäuresequenz übersetzt wird. Gendrift: Zufällige, nicht durch Selektion bewirkte Veränderung des Bestandes an Allelen (Genvarianten) einer Population. GULO: Ein Enzym (Oxidase; L-Gulonolactonoxidase), das eine chemische Vorstufe der Ascorbinsäure (Vitamin C) oxidiert. Da dieses Enzym beim Menschen fehlt, muss er Vitamin C durch Nahrung zu sich nehmen, um nicht durch Vitaminmangel zu erkranken. Homoplasie: Gleiche Sequenzanordnung in verschiedenen Organismen, die sich nicht durch gemeinsame Abstammung erklären lässt. Es handelt sich um → Konvergenz, Parallelismus oder Reversion (Rückentwicklung) auf genetischer Ebene. Hotspot: bezeichnet in der Genetik Bereiche in der DNA, bei denen vermehrt Mutationen oder Rekombinationen stattfinden. Konvergenz: gleichartige Entwicklung ähnlicher Strukturen und Funktionen einzelner Organe nicht abstammungsmäßig verwandter Organismen. Mutation: spontane oder künstlich ausgelöste Erbänderung. Nukleotid: Einzelbaustein der DNA, bestehend aus einer Stickstoff-Base, einem C5-Zucker und einem Phosphorsäurerest. Pseudogen: DNA-Abschnitt, der nicht translatiert (in eine Aminosäuresequenz übersetzt) wird, aber große Ähnlichkeit zu einem Gen hat, das tatsächlich in ein Protein übersetzt wird. Punktmutation: Eine → Mutation, bei der nur ein einziges → Nukleotid ausgetauscht, eingebaut oder herausgenommen wird.
Artübergreifende wiederkehrende Mutationen
Die heute vorliegenden biologischen Fakten legen nahe, dass Mutationen nicht nur beliebig im Genom von Organismen auftreten, sondern dass sie von molekularen Mechanismen angetrieben werden. Wäre dies ein generelles Phänomen, das bei unterschiedlichen Arten vorliegt, hätte es bedeutsame Konsequenzen für die Sichtweise von einer allgemeinen Abstammung aller Lebewesen, da Hotspots und wiederkehrende Indels (Einschübe oder Verluste) eine Illusion gemeinsamer Abstammung hervorrufen können. Die Muster von Mutationen in verschiedenen Arten von Taufliegen sind bemerkenswert ähnlich. Die Variationen, die man bei der Flügeladerung beobachtete, wurden bei etwa 21.000 Fliegen aus 117 Arten beobachtet (Houle et al. 2017). Die Forscher kamen zu dem Schluss, dass ihre Beobachtungen nicht ohne Weiteres von einem der verfügbaren Evolutionsmodelle erklärt werden können und eine wichtige Herausforderung für evolutionäre Modellierungen darstellen, da sie mit wiederkehrenden Mutationen zu tun haben.
Die Illusion ist komplett bei nicht-zufälligen Mutationen in Pseudogenen reproduktiv isolierter Arten.
Überall, wo man genauer hinschaut, begegnet einem dieses Phänomen. Auch eine detaillierte Studie der DNA-Sequenz, die für das Enzym Alkohol-Dehydrogenase (ADH4) kodiert, belegt die nicht-zufällige Häufung von Mutationen. Das ADH4-Gen ist das erste Gen, das angeschaltet wird, wenn Alkohol konsumiert wird, da es im Magen exprimiert wird. In den meisten Tieren, auch in den meisten Primaten, ist ADH4 ein langsam wirkendes Enzym und das Abbauen von Alkohol erfordert viel Zeit. In Menschen und Menschenaffen gibt es aber eine Mutation, durch die ADH4 vierzig Mal schneller Alkohol abbauen kann. Diese Mutation erlaubt es, dass man ab und zu richtig feiern kann, ohne einen 40-tägigen Kater zu bekommen. Die meisten Lebewesen werden jedoch Alkohol meiden, weil sie nicht diese Mutation haben. Es gibt aber Ausnahmen, wie zum Beispiel das Fingertier, ein mysteriöses, großäugiges Nachttier, das man nur auf Madagaskar antreffen kann. Das Fingertier – oder Aye-Aye – ist ein kleiner Primat aus der Gruppe der Lemuren, der mit seinen langen, feinfühligen Fingern Insektenlarven aus morschem Holz angelt. Merkwürdigerweise hat das Fingertier genau die gleiche Mutation in ADH4 wie Menschen und Menschenaffen. Die Mutation muss aber unabhängig entstanden sein, da es zu einem völlig anderen Zweig des evolutionären Primaten-Stammbaums gehört (Gochman et al. 2016). Falls Mutationen ausschließlich zufällig in DNA-Sequenzen auftreten würden, wären die gleichen Mutationen nicht in beiden Zweigen der Primaten zu erwarten. Diese Mutationen schaffen eine Illusion von gemeinsamer Abstammung.

Abb. 2: Variable Nukleotide des Exons X, die in den GULO (Pseudo)-Genen einer Reihe von Spezies vorhanden sind (die Nukleotiden, die in allen Organismen gleich sind, sind nicht dargestellt). Die Deletionsmutation an Position 97 (angezeigt durch *) wird normalerweise als ultimativer Beweis für die gemeinsame Abstammung zwischen Mensch und Menschenaffen interpretiert. Auf den ersten Blick mag dies ein sehr starkes Argument für eine gemeinsame Abstammung sein. Nach der Untersuchung einer großen Anzahl von Organismen, die den Ausschluss von nicht-zufälligen Mutationen ermöglichen, wird jedoch deutlich, dass die Position 97 tatsächlich ein Hotspot für nicht-zufällige Mutationen ist. (Aus dem Datensatz von Truman & Terborg 2007)
Diese Illusion ist komplett, wenn wir die nicht-zufälligen Mutationen in Pseudogenen von reproduktiv isolierten Arten betrachten. Ein in der evolutionstheoretischen Literatur ausführlich diskutiertes Beispiel sind die Mutationen im zehnten Exon* des GULO-Gens (Ohta et al. 1999). Das GULO-Gen codiert für das Protein Gulono-Lactono-Oxidase. Es katalysiert den letzten Schritt in der Biosynthese von Vitamin C. Nahezu alle Pflanzen und Tiere besitzen diesen Syntheseweg, denn Vitamin C ist unentbehrlich für die Synthese des Bindegewebes und es schützt als Antioxidans vor Alterungsprozessen. Trotzdem gibt es eine Anzahl an Tieren, die kein Vitamin C synthetisieren können. Da diese Tiere das Vitamin jedoch in ausreichender Menge mit ihrer Nahrung aufnehmen, tritt eine Vitamin-Mangelerscheinung, in diesem Fall die Avitaminose Skorbut, nicht auf. Eine große Zahl Früchte fressender Fledermäuse der Familie Chiroptera muss ohne Vitamin C auskommen. Und auch Meerschweinchen können es selbst nicht herstellen (Birney et al. 1976). Menschen und Schimpansen können Ascorbinsäure ebenfalls nicht synthetisieren. Mehr noch, alle getesteten Mitglieder der Familie der Catarrhini*, d. h. alle Menschenaffen (Schimpanse, Gorilla, Orang-Utan), können ebensowenig Vitamin C synthetisieren. Dies steht im Gegensatz zur anderen Unterordnung der Primaten, den Platyrrhini, bei denen Vitamin C in der Leber synthetisiert wird. Der Verlust der Fähigkeit, Vitamin C zu synthetisieren, ist offensichtlich häufig im Tierreich vorgekommen und Teile des inaktiven GULO-Gens sind in mehreren Arten noch immer identifizierbar. Dass ein defektes GULO-Gen sich durch Gendrift* verbreitete, ist eine Möglichkeit, aber eher unwahrscheinlich, wenn die Populationen der oben erwähnten Organismen groß waren. Erst wenn alle diese Organismen einen genetischen Engpass durchlaufen hätten, könnte das defekte Gen in der gesamten Population fixiert werden. Interessanterweise gibt es Hinweise, dass alle Tierarten einen solchen Engpass durchlaufen haben (Terborg 2019).
Der wichtigste Grund für die Faszination des GULO-Gens ist die Tatsache, dass eine ähnliche Deletion*, wie sie auf Position 97 des zehnten Exons* – ExonX – im Menschen angetroffen wird, auch in Schimpansen, Orang-Utans und Makaken vorkommt. Es ist selbstverständlich, dass diese Deletion von Evolutionstheoretikern als endgültiger Beweis für gemeinsame Abstammung von Menschen und Affen interpretiert wird. Und es wäre tatsächlich ein starkes Indiz, wenn Mutationen ausschließlich ein zufälliges und unvorhersehbares Phänomen wären. Die in allen Catharrhini vorhandenen Deletionen könnten aber auch die Folge eines artübergreifenden Hotspots sein, einer genetischen Stelle, an der sich Mutationen bevorzugt ansammeln. In Kombination mit dem oben erwähnten Engpass wäre es dann möglich, dass diese Organismen dieselbe Deletion unabhängig voneinander erhielten. Diese Hypothese muss natürlich untermauert werden. Denn sollte es sich herausstellen, dass sie zutrifft oder es zumindest gute Argumente für sie gibt, wäre die molekulare Beweisführung für gemeinsame Abstammung fragwürdig.
Die veröffentlichten Sequenzanalysen des GULO-Gens wurden alle mit einer kleinen Anzahl von Spezies durchgeführt (Ohta et al. 1999, Nishikimi et al. 1992; 1994, Inai et al. 2003). Studien mit wenigen Arten sind jedoch, wie oben schon angemerkt, nicht geeignet, artübergreifende Hotspots zu identifizieren. Deshalb haben wir 2009 eine eigene Studie durchgeführt, durch die wir einen nicht-zufälligen Hotspot an Position 97 ausschließen wollten (Truman & Terborg 2007). Abb. 2 zeigt die DNA-Sequenz des zehnten Exons des GULO-Gens (Exon X), wie sie in 11 verschiedenen Organismen vorkommt. Beim Vergleich der Sequenzen ist sofort ersichtlich, dass mehrere Positionen Hotspots für Mutationen sind, darunter die Positionen 76 und 131. Aber auch die Deletion auf Position 97 ist ein Hotspot für Mutationen. Es ist auffällig, dass wir hier in allen untersuchten Organismen einen anderen DNA-Buchstaben auf dieser Position antreffen. Wenn wir in Abb. 2 Position 97 von unten nach oben betrachten, lesen wir A-C-G-A-C-A-G. Der Vergleich der angrenzenden Positionen 96 und 98 zeigt immer G-G-G-G-G-G-G; es sind also sehr stabile, unveränderliche Positionen. Position 97 dagegen ist eine sehr variable Position. Von allen DNA-Buchstaben verändert sich Position 97 am häufigsten. Position 97 ist eine sehr instabile Stelle in einer DNA-Sequenz, an der Mutationen vorzugsweise auftreten. Wir konnten die Null-Hypothese, dass es sich hier um einen artübergreifenden Hotspot handelt, somit nicht ausschließen. Die Deletion kann sehr gut als artübergreifender Hotspot betrachtet werden. In Primaten manifestiert sich diese Instabilität als eine Deletion und erzeugt die Illusion einer gemeinsamen Abstammung (Abb. 2). Dass artübergreifende wiederkehrende Mutationen existieren, belegt nicht nur das GULO-Gen, sondern auch das Uratoxidase-Gen, wo sie auch die Illusion einer gemeinsamen Abstammung erwecken (Oda et al. 2002).
Homoplasie
Artübergreifende Hotspots verdienen besondere Aufmerksamkeit, weil sie auf elegante Weise ein überraschendes Resultat der molekularen Phylogenetik erklären: Homoplasie*. Evolutionstheoretiker sprechen von Homoplasie, wenn DNA-Sequenzen in Organismen, die in einem evolutionären Stammbaum nicht eng verwandt sind, identisch sind (Abb. 3):
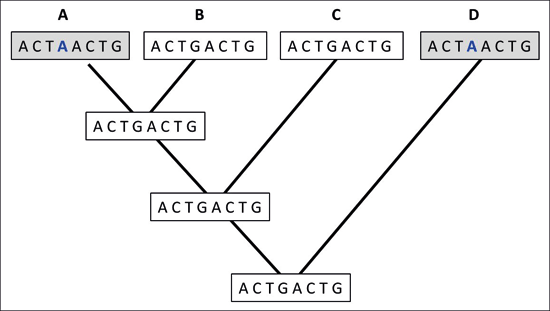
Abb. 3: In der Evolutionsbiologie bezieht sich der Begriff „Homoplasie“ auf ein gemeinsames Merkmal oder eine gemeinsame Merkmalsausprägung, die aufgrund dieser Gemeinsamkeit evolutionäre Verwandtschaft nahezulegen scheint, in Wirklichkeit aber unabhängig erworben oder verloren wurde. Bei unabhängigem Erwerb spricht man auch von Konvergenz. In der Abbildung scheinen die DNA-Sequenzen der Organismen A und D am engsten miteinander verwandt zu sein, obwohl die Abstammungslinien die Organismen am weitesten voneinander entfernt bestimmen (z. B.: A = Mensch; B = Schimpanse; C = Gorilla; D = Makake). Solche Konvergenzen rufen eine Illusion der gemeinsamen Abstammung hervor.
„Homoplasie wird in der theoretischen Phylogenetik seit langem anerkannt, und man hat sich viel Mühe gegeben, ihre Ursachen zu verstehen und entsprechende Korrekturen vorzunehmen. Die beobachteten Muster [...] geben jedoch Anlass zur Sorge, dass das Ausmaß der Homoplasie viel größer sei als bei allgemein akzeptierten Modellen der Sequenzevolution erwartet und dass die damit verbundenen Konsequenzen für die Grenzen der phylogenetischen Auflösung nicht ausreichend gewürdigt werden“ (Rokas & Carroll 2006).5
Eine neuere genetische Analyse brachte beispielsweise ans Licht, dass die meisten Mutationen, die im Menschen degenerative Krankheiten verursachten, auch bei Rhesusaffen angetroffen werden, im Gegensatz zu den Erwartungen jedoch nicht beim Schimpansen. Von den 229 Mutationen, die der Mensch und der Rhesusaffe gemeinsam hatten, wurden nur 97 beim Schimpansen aufgefunden (Rokas 2006; Rhesus Macaque Genome Sequencing and Analysis Consortium 2007). Der Mensch gleicht in diesem Fall dem Rhesusaffen mehr als dem Schimpansen.6 Vergleichende Analysen zeigten, dass ein großer Teil der Gene des Menschen den Gorilla-Genen mehr ähnelt als denen des Schimpansen, was die Annahme von Homoplasien erfordert. Aufgrund des gegenwärtigen biologischen Wissens können Mutationen nicht mehr ausschließlich als zufällige genetische Veränderungen betrachtet werden, die nicht vorhergesagt werden können. Im Gegenteil: Wenn wir genügend Sequenzdaten von möglichst vielen Probanden haben, können wir mit hoher Wahrscheinlichkeit vorhersagen, wo in bestimmten Sequenzen Mutationen auftreten werden. Da sich Mutationen nicht nur zufällig in einer DNA-Sequenz ansammeln, besitzen wir nun auch eine plausible biologische Erklärung für Homoplasien: Homoplasie wird durch artübergreifende wiederkehrende Mutationen hervorgerufen. Evolutionstheoretiker dagegen sind der Auffassung, dass Homoplasien die bekannten Abstammungslinien stören. Es könnte aber durchaus sein, dass sie ausgehend vom Darwin‘schen Paradigma auf das falsche Pferd gesetzt haben. Man geht von vornherein davon aus, dass gemeinsame Abstammung wahr ist und dass Mutationen nur zufällig in den Sequenzen auftreten – mit Ausnahme einiger bekannter Hotspots. Beide Annahmen haben sich jedoch als falsch erwiesen.
Wenn gleichartige Mutationen auch nicht-zufällig in einer DNA-Sequenz auftreten können, lassen sich die genetischen Daten in einem Schöpfungsrahmen interpretieren.
Die Experten schätzen, dass 10-15 % der Mutationen auf Homoplasie zurückzuführen sind (Rokas & Carroll 2006). Da Homoplasie den nicht-zufälligen Charakter von Mutationen widerspiegeln könnte, schafft sie ein offensichtliches Dilemma: Wie können wir zwischen Homologie und Homoplasie unterscheiden, also zwischen Mutationen, die zur Ermittlung einer gemeinsamen Abstammung genutzt werden können, und solchen, bei denen das nicht der Fall ist? Die Antwort ist: Wir können es nicht. Es kann durchaus sein, dass die in der Evolutionsliteratur vorgestellten makroevolutionären Stammbäume eine Illusion sind, die durch eine Kombination von gemeinsamem Sequenz-Design und artübergreifenden Hotspots erzeugt werden. Für diese Erklärung spricht, dass eine gemeinsame Abstammung von Mensch und Schimpanse durch die neusten genetischen Befunde sehr disputabel ist. Der genetische Unterschied zwischen beiden Organismen wird heutzutage auf 16 % bestimmt und umfasst mehrere hundert Gene und eine Fülle neuer genetischer Information, für deren Ursprung es in einer naturalistischen Weltsicht keine plausible Erklärung gibt. Diese neue, einzigartige biologische Information, die wir jeweils im Genom von Mensch und Tier vorfinden, ist ein klares Gegenargument zur These der universellen Abstammung.
Bestimmte molekulargenetische Daten wie mutmaßlich vererbte Fehler schienen bisher ein starkes Argument gegen eine getrennte Erschaffung von Grundtypen zu sein. Mit der Erkenntnis, dass sich gleichartige Mutationen auch nicht-zufällig und unabhängig in einer DNA-Sequenz anhäufen können, wird dieses Argument entkräftet und die genetischen Daten lassen sich somit nach wie vor in einem Schöpfungsrahmen interpretieren: Die artübergreifenden Mutationen, die eine Illusion gemeinsamer Abstammung erwecken, können als Konvergenzen verstanden werden.
1 Man hatte zwar festgestellt, dass ,,hot-spots“ in den DNA-Sequenzen vorkamen, aber man unterstellte, dass sie keinen nennenswerten Einfluss auf die Abstammungslinien ausüben würden.
2 Original: „Mutation is random in two senses. First, although we may be able to predict the probability that a certain mutation will occur, we cannot predict which of a large number of gene copies will undergo the mutation. Second, and more importantly, mutation is random in the sense that the chance that a particular mutation will occur is not influenced by whether or not the organism is in an environment in which that mutation would be advantageous.”
3 Dabei geht man davon aus, dass solche Mutationen zufälligerweise an den gleichen Stellen auftraten, aber durch Selektion fixiert wurden, weil sie den Organismen einen reproduktiven Vorteil verschafften.
4 Die verborgenen Sequenzen werden als „dunkle DNA“ bezeichnet in Anspielung auf die dunkle Materie im Kosmos, die wir nicht entdecken können, obwohl sie 25 % des Universums ausmachen sollte. Im Gegensatz zu dieser kosmischen dunklen Materie wird die dunkle DNA nicht wirklich vermisst.
5 Original: „Homoplasy has long been appreciated in theoretical phylogenetics, with much effort invested into understanding its causes and providing corrections for them. However, the observed patterns … give cause for concern that the extent of homoplasy is much greater than expected under widely accepted models of sequence evolution and that the attendant consequences for the limits to phylogenetic resolution are not sufficiently appreciated.“
6 Die Voreingenommenheit der Darwin‘schen Theoretiker rührt von der Tatsache her, dass nicht berücksichtigt wird, dass genetische Übereinstimmungen zwischen Mensch und Schimpanse auch durch Konvergenz bzw. Homoplasie verursacht werden können. Daher werden sie als Beweise für gemeinsame Abstammung interpretiert.
Human adaptation to arsenic in Andean populations of the Atacama Desert. Am. J. Phys. Anthropol. 163,192–199; doi: 10.1002/ajpa.23193.
Das kooperative Gen. Hamburg: Hoffmann und Campe.
Inability of bats to synthesise L-ascorbic acid. Nature 260, 626-628.
Darwin Revisited, Scholars Press.
Darwin in the Genome. The McGraw Hill Companies.
Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nature Biotechnology 34, 155–163.
Natural radioactivity and human mitochondrial DNA mutations. Proc. Natl. Acad. Sci. 99, 13950–13954,
Evolutionary Biology, 3rd ed.,Sinauer Associates, Sunderland, MA, p. 178-179.
Alcohol discrimination and preferences in two species of nectar-feeding primate. The Royal Society.
Introducing ‘dark DNA’ – the phenomenon that could change how we think about evolution. http://theconversation.com/introducing-dark-dna-the-phenomenon-that-could-change-how-we-think-about-evolution-82867
Identification of a novel locus associated with skin colour in African-admixed populations. Sci. Rep. 7:44548.
Mutation predicts 40 million years of fly wing evolution. Nature, doi:10.1038/nature23473.
The whole structure of the human non-functional L-gulono-gamma-lactone oxidase gene – the gene responsible for scurvy – and the evolution of repetitive sequences thereon J. Nutr. Sci. Vitaminol. 49, 315–319.
The origin of spontaneous mutations: specificity and directionality of base substitution, frameshift, and sequence-substitutions mutageneses. Annu. Rev. Genet. 39, 297–303.
Guinea pigs possess a highly mutated gene for L-gulono-gamma-lactone oxidase, the key enzyme for l-ascorbic acid biosynthesis missing in this species. J. Biol. Chem. 267, 21967–21972.
Cloning and chromosomal mapping of the human non-functional gene for L-gulono-gamma-lactone oxidase, the enzyme for l-ascorbic acid biosynthesis missing in man. J. Biol Chem. 269, 13685–13688.
Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol. Biol. Evol.19, 640-653.
Random nucleotide substitutions in primate nonfunctional gene for L-gulono-gamma-lactone oxidase, the missing enzyme in L-ascorbic acid biosynthesis, Biochim. Biophys. Acta 1472, 408–411.
AID/APOBEC deaminases and cancer. Oncoscience 28, 320-333.
Evolutionary and Biomedical Insights from the Rhesus Macaque Genome, Science 316, 222–234.
Bushes in the tree of life, PLoS Biol. 4(11):e352.
Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303, 223-236.
A screen for fast evolving genes from Drosophila, Proc. Natl. Acad. Sci. 94, 9746–9750.
mtDNA control-region sequence variation suggests multiple independent origins of an ‘Asian-specific’ 9-bp deletion in sub-Saharan Africans, Am. Hum. Gen. 58, 595-608.
Not by Chance.The Judaica Press Ltd.
Überraschende genetische Engpässe – Hinweis auf eine globale Dezimierung der Tierwelt? Stud. Integr. J. 26, 39–42.
Why the shared mutations in the Hominidae exon X GULO pseudogene are not evidence for common descent. J. Creat. 21.
DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science. 363, 81-84.
Themen | Kurzbeiträge | Streiflichter
Studiengemeinschaft WORT und WISSEN e.V.
Letzte Änderung: 6/21/21
Webmaster