
Evolution „täuschte“ Wissenschaftler über 100 Jahre lang
Downloads
Evolution „täuschte“ Wissenschaftler über 100 Jahre lang
Schon zur Zeit Darwins war der Stammbaum aller Lebewesen das Herz der Evolutionslehre. Das einzige Schaubild, das in Darwins „Origin of Species“ enthalten war, stellt das Prinzip der gemeinsamen Abstammung von Lebewesen schematisch dar. Aufgestellt wurden Stammbäume anhand von gestaltlichen Ähnlichkeiten (Morphologie) der Lebewesen. Dabei wird der Grad der Ähnlichkeiten als Grad der stammesgeschichtlichen Verwandtschaft interpretiert. Anhand solcher intuitiv leicht erfassbaren Schaubilder konnte die Evolutionslehre den Menschen vermittelt werden. Evolution wurde so zum nicht mehr hinterfragten Kerninhalt des Biologiestudiums an vielen Universitäten rund um die Welt.
Doch inzwischen wendet sich das Blatt und es sieht so aus, als ob „jahrhundertelange wissenschaftliche Arbeiten revidiert werden müssen“ (University of Bath 2022). Stammbäume werden seit einigen Jahrzehnten zunehmend durch den Vergleich genetischer Daten oder Aminosäureabfolgen in Proteinen verschiedener Lebewesen aufgestellt. Entgegen der voraussagenden Annahme von Ernst Mayr, dem „Darwin des 20. Jahrhunderts“, ergeben sich jedoch in weiten Teilen völlig andere Abstammungszusammenhänge, wenn molekulare Daten anstelle gestaltlicher Merkmale für das Aufstellen von Stammbäumen verwendet werden.
Dem Vergleich der beiden Methoden widmete eine Gruppe britischer Evolutionsbiologen um Jack Oyson eine Studie. Sie verglichen 48 evolutionäre Stammbäume, die jeweils anhand von morphologischen und molekularen Daten aufgestellt wurden. Dabei zeigten sich große Unterschiede zwischen den molekularen und morphologischen Stammbäumen. Um nur zwei von vielen Beispielen zu nennen: Raubtiere (Carnivora) und Schuppentiere (Pholidota) sind nach morphologischen Kriterien stammesgeschichtlich sehr weit voneinander entfernt, aber nach molekularen Kriterien sehr nah verwandt. Das Umgekehrte gilt für Insektenfresser (Eulityphla) und Tenrekartige (Afrosoricida). Diese Diskrepanzen sind nicht neu, scheinen aber riesige Ausmaße zu haben: „Beispielsweise haben phylogenomische Analysen von Plazentasäugetieren die Abfolge der tiefen Verzweigungen, die traditionell durch die Morphologie gestützt wurden, drastisch verändert“ (Oyson et al. 2022).
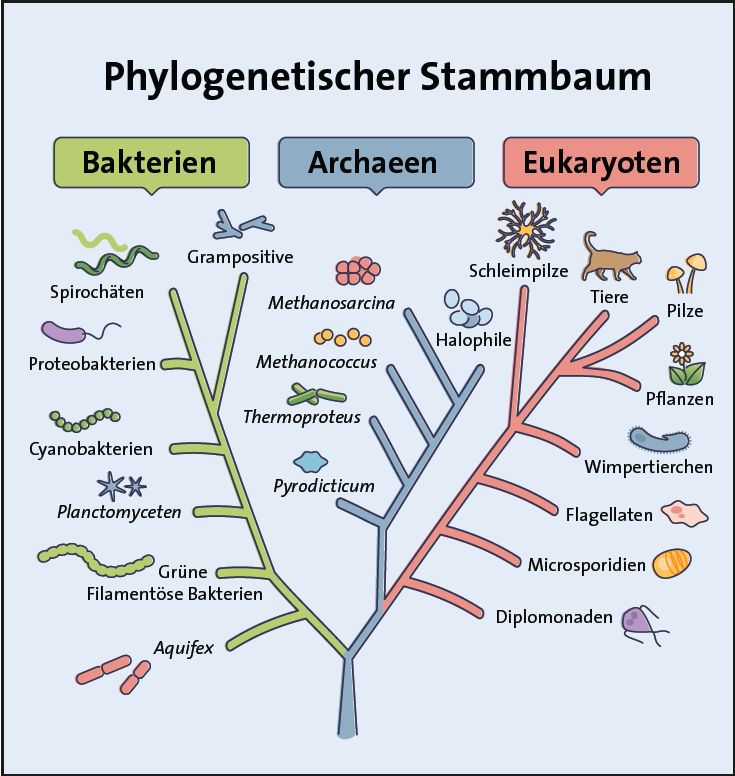
Figure 1. Abb. 1 Phylogenetische Stammbäume sind ein Kerninhalt des heutigen Biologiestudiums. Links: Stammbaum des Lebens nach Ernst Haeckel, rechts: derzeit akzeptierter evolutionärer Stammbaum aller Lebewesen. (VectorMine, AdobeStock)
In der Studie wurde zudem geprüft, inwiefern die etablierten Abstammungsverhältnisse mit der biogeographischen Verteilung der Lebewesen übereinstimmen. Die Annahme ist hier, dass eine nahe Verwandtschaft verschiedener Arten auch mit einer geringen Entfernung der jeweiligen Habitate einhergeht. Es ergab sich, dass die molekularen Stammbäume etwas besser zur biogeographischen Verteilung passten als die morphologischen. Der Unterschied war dabei nicht groß: Im Vergleich zu zufälligen Verteilungen der Lebewesen ist diejenige nach morphologischen Stammbaumkriterien um 54 % besser, diejenige nach molekularen Daten um 65 %. Die Forschungsgruppe fand allerdings auch Familien, für deren Stammbäume die morphologischen Daten besser zur biogeographischen Verteilung passen: Hunde (Canidae), Eichhörnchen (Sciuridae), Fledermäuse (Chiroptera) und Kängurus (Macropodidae).
Es blieb nicht unerwähnt, dass molekulare Stammbäume wesentlich häufiger „Homologieprobleme“ aufweisen, d. h. es ergeben sich bei sehr weit voneinander entfernten Organismen unerwartet stark ausgeprägte Ähnlichkeiten. Dieser Sachverhalt wird inzwischen damit erklärt, dass die Evolution auf rätselhafte Weise in voneinander unabhängigen Abläufen wiederholt zu den gleichen biologischen Bauplänen und Organen gelangt. Auch wenn das mit einer richtungs- und ziellos verlaufenden Evolution in keiner Weise zusammenpasst, wird es inzwischen für ein häufiges Phänomen gehalten.
Der an der Arbeit beteiligte Matthew A. Wills äußerte sich dazu wie folgt: „Neuerdings sehen wir anhand der molekularen Daten, dass konvergente Evolution ständig geschieht. Dinge, von denen wir dachten, sie seien nah verwandt, sind offenbar auf dem Stammbaum des Lebens weit voneinander entfernt“ (University of Bath 2022).
Es findet also eine Neuinterpretation alter Daten in großem Maßstab statt, und das, was früher einmal als eine „Homologie“ (also als Merkmal gemeinsamer Abstammung) galt, wird jetzt oft als „Konvergenz“ angesehen. Dabei legen die Forscher eine überraschende Sicherheit an den Tag – ähnlich wie frühere Generationen in Bezug auf vermeintliche „Homologien“. Wills sagte dazu: „Das bedeutet, dass konvergente Evolution uns verschaukelt hat – selbst die schlauesten Biologen und Anatomen – über 100 Jahre lang“ (University of Bath 2022).
Dabei ist den Autoren der Studie durchaus bewusst, dass auch molekulare Stammbäume alles andere als sicher sind: „In gleicher Weise gibt es Konflikte zwischen Stammbäumen, die auf molekularen Daten beruhen, insbesondere wenn sie von unterschiedlichen genetischen Einheiten hergeleitet werden“ (Oyson et al. 2022).
Eine evolutionäre Systematik der Lebewesen scheint in immer weitere Ferne zu rücken, je mehr geforscht wird. Ausgehend vom Grundtypenmodell der Schöpfungslehre würden sich viele der angeführten Widersprüche dagegen auflösen, weil in diesem Rahmen von einer freien Kombinierbarkeit von Merkmalen ausgegangen werden kann.
[Oyson JW et al. (2022) Molecular phylogenies map to biogeography better than morphological ones. Commun. Biol. 5, 521, https://doi.org/10.1038/s42003-022-03482-x • University of Bath (2022) Study suggests that most of our evolutionary trees could be wrong. Pressemitteilung, https://phys.org/news/2022-06-evolutionary-trees-wrong.html] B. Schmidtgall
Evolution „täuschte“ Wissenschaftler über 100 Jahre lang
Schon zur Zeit Darwins war der Stammbaum aller Lebewesen das Herz der Evolutionslehre. Das einzige Schaubild, das in Darwins „Origin of Species“ enthalten war, stellt das Prinzip der gemeinsamen Abstammung von Lebewesen schematisch dar. Aufgestellt wurden Stammbäume anhand von gestaltlichen Ähnlichkeiten (Morphologie) der Lebewesen. Dabei wird der Grad der Ähnlichkeiten als Grad der stammesgeschichtlichen Verwandtschaft interpretiert. Anhand solcher intuitiv leicht erfassbaren Schaubilder konnte die Evolutionslehre den Menschen vermittelt werden. Evolution wurde so zum nicht mehr hinterfragten Kerninhalt des Biologiestudiums an vielen Universitäten rund um die Welt.
Doch inzwischen wendet sich das Blatt und es sieht so aus, als ob „jahrhundertelange wissenschaftliche Arbeiten revidiert werden müssen“ (University of Bath 2022). Stammbäume werden seit einigen Jahrzehnten zunehmend durch den Vergleich genetischer Daten oder Aminosäureabfolgen in Proteinen verschiedener Lebewesen aufgestellt. Entgegen der voraussagenden Annahme von Ernst Mayr, dem „Darwin des 20. Jahrhunderts“, ergeben sich jedoch in weiten Teilen völlig andere Abstammungszusammenhänge, wenn molekulare Daten anstelle gestaltlicher Merkmale für das Aufstellen von Stammbäumen verwendet werden.
Dem Vergleich der beiden Methoden widmete eine Gruppe britischer Evolutionsbiologen um Jack Oyson eine Studie. Sie verglichen 48 evolutionäre Stammbäume, die jeweils anhand von morphologischen und molekularen Daten aufgestellt wurden. Dabei zeigten sich große Unterschiede zwischen den molekularen und morphologischen Stammbäumen. Um nur zwei von vielen Beispielen zu nennen: Raubtiere (Carnivora) und Schuppentiere (Pholidota) sind nach morphologischen Kriterien stammesgeschichtlich sehr weit voneinander entfernt, aber nach molekularen Kriterien sehr nah verwandt. Das Umgekehrte gilt für Insektenfresser (Eulityphla) und Tenrekartige (Afrosoricida). Diese Diskrepanzen sind nicht neu, scheinen aber riesige Ausmaße zu haben: „Beispielsweise haben phylogenomische Analysen von Plazentasäugetieren die Abfolge der tiefen Verzweigungen, die traditionell durch die Morphologie gestützt wurden, drastisch verändert“ (Oyson et al. 2022).
Figure 1. Abb. 1 Phylogenetische Stammbäume sind ein Kerninhalt des heutigen Biologiestudiums. Links: Stammbaum des Lebens nach Ernst Haeckel, rechts: derzeit akzeptierter evolutionärer Stammbaum aller Lebewesen. (VectorMine, AdobeStock)
In der Studie wurde zudem geprüft, inwiefern die etablierten Abstammungsverhältnisse mit der biogeographischen Verteilung der Lebewesen übereinstimmen. Die Annahme ist hier, dass eine nahe Verwandtschaft verschiedener Arten auch mit einer geringen Entfernung der jeweiligen Habitate einhergeht. Es ergab sich, dass die molekularen Stammbäume etwas besser zur biogeographischen Verteilung passten als die morphologischen. Der Unterschied war dabei nicht groß: Im Vergleich zu zufälligen Verteilungen der Lebewesen ist diejenige nach morphologischen Stammbaumkriterien um 54 % besser, diejenige nach molekularen Daten um 65 %. Die Forschungsgruppe fand allerdings auch Familien, für deren Stammbäume die morphologischen Daten besser zur biogeographischen Verteilung passen: Hunde (Canidae), Eichhörnchen (Sciuridae), Fledermäuse (Chiroptera) und Kängurus (Macropodidae).
Es blieb nicht unerwähnt, dass molekulare Stammbäume wesentlich häufiger „Homologieprobleme“ aufweisen, d. h. es ergeben sich bei sehr weit voneinander entfernten Organismen unerwartet stark ausgeprägte Ähnlichkeiten. Dieser Sachverhalt wird inzwischen damit erklärt, dass die Evolution auf rätselhafte Weise in voneinander unabhängigen Abläufen wiederholt zu den gleichen biologischen Bauplänen und Organen gelangt. Auch wenn das mit einer richtungs- und ziellos verlaufenden Evolution in keiner Weise zusammenpasst, wird es inzwischen für ein häufiges Phänomen gehalten.
Der an der Arbeit beteiligte Matthew A. Wills äußerte sich dazu wie folgt: „Neuerdings sehen wir anhand der molekularen Daten, dass konvergente Evolution ständig geschieht. Dinge, von denen wir dachten, sie seien nah verwandt, sind offenbar auf dem Stammbaum des Lebens weit voneinander entfernt“ (University of Bath 2022).
Es findet also eine Neuinterpretation alter Daten in großem Maßstab statt, und das, was früher einmal als eine „Homologie“ (also als Merkmal gemeinsamer Abstammung) galt, wird jetzt oft als „Konvergenz“ angesehen. Dabei legen die Forscher eine überraschende Sicherheit an den Tag – ähnlich wie frühere Generationen in Bezug auf vermeintliche „Homologien“. Wills sagte dazu: „Das bedeutet, dass konvergente Evolution uns verschaukelt hat – selbst die schlauesten Biologen und Anatomen – über 100 Jahre lang“ (University of Bath 2022).
Dabei ist den Autoren der Studie durchaus bewusst, dass auch molekulare Stammbäume alles andere als sicher sind: „In gleicher Weise gibt es Konflikte zwischen Stammbäumen, die auf molekularen Daten beruhen, insbesondere wenn sie von unterschiedlichen genetischen Einheiten hergeleitet werden“ (Oyson et al. 2022).
Eine evolutionäre Systematik der Lebewesen scheint in immer weitere Ferne zu rücken, je mehr geforscht wird. Ausgehend vom Grundtypenmodell der Schöpfungslehre würden sich viele der angeführten Widersprüche dagegen auflösen, weil in diesem Rahmen von einer freien Kombinierbarkeit von Merkmalen ausgegangen werden kann.
[Oyson JW et al. (2022) Molecular phylogenies map to biogeography better than morphological ones. Commun. Biol. 5, 521, https://doi.org/10.1038/s42003-022-03482-x • University of Bath (2022) Study suggests that most of our evolutionary trees could be wrong. Pressemitteilung, https://phys.org/news/2022-06-evolutionary-trees-wrong.html] B. Schmidtgall